Новые успехи в предсказании пространственной структуры белков
19 октября 2007
Новые успехи в предсказании пространственной структуры белков
- 2216
- 0
- 3
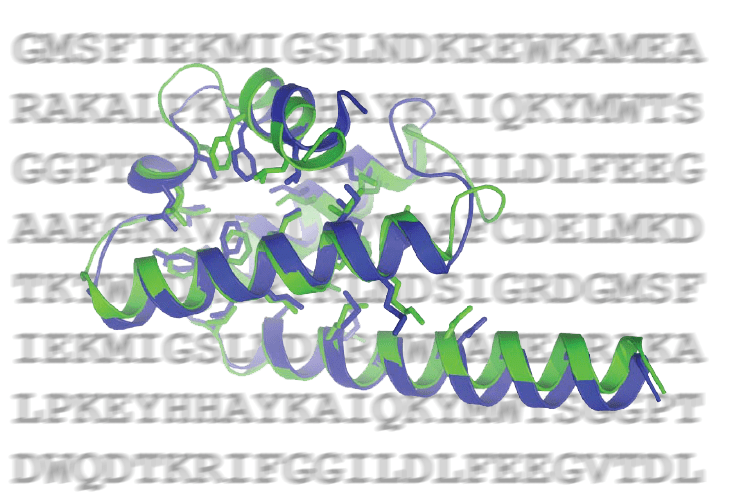
Благодаря новой методике учёным удалось весьма точно предсказать структуру бактериального белка с неизвестной функцией (зелёный — предсказание, синий — появившаяся позднее кристаллографическая структура)
-
Автор
-
Редакторы
Теоретическое предсказание пространственной структуры белков — одна из наиболее актуальных задач структурной биологии, поскольку её получение в прямом эксперименте далеко не всегда возможно. В результате распределённого компьютерного эксперимента с участием более 70 000 пользователей персональных компьютеров по всему миру впервые с высокой точностью предсказана структура небольшого глобулярного белка, руководствуясь лишь его аминокислотной последовательностью. Учёные отмечают, что получаемые модели, кроме всего прочего, могут быть использованы для уточнения структур белков, получаемых экспериментально.
Пространственная структура белков — ключ к пониманию их биологической функции и механизма работы. Знание строения «молекулярных машин» может дать учёным незаменимую подсказку для направленного дизайна новых, эффективных и безопасных лекарственных соединений [1]. Наиболее часто используемый метод определения трёхмерной структуры белка — рентгеновская кристаллография, позволяющая по дифракционной картине, получающейся от рассеяния рентгеновских лучей на кристаллах белка, восстановить трёхмерную карту электронной плотности и пространственную структуру молекулы. Однако далеко не во всех случаях желаемую структуру можно определить за фиксированное время и уложиться в скромный бюджет — так сложен и длителен кристаллографический эксперимент, — и в этих случаях для теоретического предсказания строения белка используют компьютерные методы.
В последние годы с увеличением возможностей вычислительной техники продолжают совершенствоваться и методы предсказания структуры белка, причём наибольшее внимание уделяется тем подходам, которые способны предсказать требуемую структуру «из первых принципов» (то есть, не используя никаких дополнительных «вводных» данных, кроме аминокислотной последовательности моделируемого белка). В случае же, если у интересующего белка есть близкий «родственник», структура которого уже известна, задача сильно упрощается: из-за схожести структур близкородственных белков модель можно построить как бы «по шаблону» с уже известным белком. Однако для белков, не имеющих близких структурных гомологов, теоретическое предсказание строения является очень трудноразрешимой задачей.
Дэвид Бэйкер (David Baker), учёный из Медицинского института имени Ховарда Хьюза (Howard Hughes Medical Institute, HHMI), и его коллеги из университетов Вашингтона и Кембриджа, докладывают в журнале Nature [2] о существенном прогрессе в предсказании трёхмерных структур белков. Одним из наиболее ярких результатов их работы стало предсказание с недостижимой ранее точностью строения одного из белков бактерии Bacillus halodurans, исходя исключительно из его первичной последовательности, составляющей 112 аминокислотных остатков. (Кристаллографическая структура этого белка, по сравнению с которой и определяется «точность», стала доступна уже после построения модели.)
«Это, безусловно, наиболее значимый результат из описанных в нашей статье, — говорит Бэйкер. — В случае этого белка всё, что мы знали — это его последовательность: ни ЯМР-данными, ни структурами родственных белков мы не располагали. Имея только последовательность, мы построили модели и, выбрав из них низкоэнергетические, мы обнаружили, что они очень точны. Это первый случай, когда структуру глобулярного белка удалось предсказать безо всяких дополнительных допущений» [3].
Основой всех современных алгоритмов предсказания структуры белков является поиск структуры с наименьшей потенциальной энергией, что и представляет из себя сложнейшую вычислительную задачу из-за астрономического количества степеней свободы, которыми обладает молекула белка. «Мы можем получить структуру, очень близкую к правильной, но всё же не совсем, — говорит Дэвид Бэйкер. — Вы могли бы подумать, что достаточно лишь „потрясти“ эту структуру туда-сюда на компьютере, и получишь то, что нужно. Однако чаще всего энергетические барьеры слишком велики и белок просто „застревает“ в какой-то одной форме».
Алгоритм, реализованный в их программе для предсказания структуры белков Rosetta@Home, чрезвычайно ресурсоёмок — он основан на многократном повторении циклов генерации ансамблей моделей, оптимизации их потенциальной энергии в полноатомном силовом поле и идентификации наиболее вероятных «ошибочных» участков с последующим направленным «перестраиванием». Для получения необходимого машинного времени расчеты параллельно производились на более чем 70 000 персональных компьютерах по всему миру (для этого желающим «поучаствовать» в эксперименте предлагалось установить на свой компьютер программу-клиент), а также на суперкомпьютере IBM Blue Gene/L, установленном в Национальной лаборатории Аргон США.
Стратегия, предложенная Бэйкером, называется «направленное перестраивание и оптимизация» (“targeted rebuilding and refining”) и заключается в том, что наиболее структурно «нестабильные» участки молекулы (широко варьирующие от модели к модели в ансамбле структур-предсказаний) считаются ошибочными и перестраиваются заново с помощью алгоритма глобальной оптимизации. Этот алгоритм временно «размыкает» полипептидную цепь, что позволяет более пóлно исследовать конформационное пространство данного участка. «Это как если у вас есть клубок верёвки, часть которой скручивается как-то не так, как надо, — пытается популярно объяснить суть своего метода Дэвид. — Вы просто вырезаете его и исследуете возможные конформации на компьютере до тех пор, пока он не начнёт скручиваться так, как вы ожидаете». Алгоритм повторяется раунд за раундом с целью провести селекцию моделей с наиболее низкой потенциальной энергией, чем-то напоминая естественный отбор.
Структурный биолог Элеанор Додсон (Eleanor Dodson) в новостной колонке Nature [4] подчёркивает значимость работы Дэвида Бэйкера и его коллег: «Этот поистине революционный подход прогрессивен по всем фронтам: привлечение невероятных вычислительных ресурсов, использование известных пространственных структур, разработка новых алгоритмов поиска связей структура–последовательность и постоянно улучшающаяся способность нахождения низкоэнергетичских конформаций молекулы. В будущем этот подход, несомненно, даст ценную структурную информацию об объектах, которые трудно исследовать экспериментально.»
Однако успех, достигнутый исследователями, имеет свои пределы: дело в том, что такой высокой точности предсказания удалось достичь лишь в одном случае из 26, описанных в работе. Ещё в семи случаях предсказанная модель, хоть и оказывалась не очень точной, всё же была «ближе» к реальной структуре, чем наиболее близкий гомолог из числа известных — что тоже является неплохим результатом. Впрочем, даже если предсказание структуры «с нуля» оказывается не таким успешным, работа не пропадает зря: учёные показали, что предложенный ими алгоритм может эффективно использоваться для оптимизации структур, полученных с помощью метода спектроскопии ядерного магнитного резонанса (ЯМР), часто недостаточно точных по сравнению с кристаллографическими. Кроме того, этот подход способен стать существенным подспорьем и в рентгеновской кристаллографии.
Дело в том, что расшифровка дифракционной картины требует решения задачи фаз для дифракционных пиков, которая существенно облегчается, если доступна пространственная структура какого-нибудь родственного белка (проводимая при этом процедура, предназначенная для оценки фаз, называется изоморфным замещением). Причём ранее было отмечено, что «качество» используемой «подспудной» модели должно быть очень высоким: так, например, структуры, определённые с помощью ЯМР-спектроскопии или традиционные теоретические модели, основанные на гомологии с родственными белками, не подходят для решения задачи фазирования. Их работа, считает Бэйкер, может дать начало новому приёму в кристаллографии — полностью компьютерному (in silico) расчету фаз из теоретических моделей, что существенно облегчит задачу получения структуры во многих случаях.
«Главное, что можно подметить из нашей работы, это то, что предсказание структуры белков (по крайней мере, небольших) — уже достаточно зрелая область, чтобы применяться для уточнения ЯМР-структур, а также теоретических моделей белков, полученных другими способами, — говорит Бэйкер. — А в особо удачных случаях можно рассчитывать и на то, что хорошая модель получится из одной только последовательности» [3].
«Проект Rosetta@home уникален не только с точки зрения научных достижений — вокруг него возникло научно-образовательное сообщество. Людям было интересно, что за вычисления производит их компьютер, и они начинали интересоваться структурой белков и в целом — молекулярной биологией», — добавил он.
Литература
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Bin Qian, Srivatsan Raman, Rhiju Das, Philip Bradley, Airlie J. McCoy, et. al.. (2007). High-resolution structure prediction and the crystallographic phase problem. Nature. 450, 259-264;
- New approach builds better proteins inside a computer. (2007). ScienceDaily;
- Eleanor J. Dodson. (2007). Protein predictions. Nature. 450, 176-177.



Комментарии
0Чтобы оставить комментарий, необходимо
войти