
Код жизни: прочесть не значит понять
02 декабря 2010
Код жизни: прочесть не значит понять
- 20581
- 2
- 16

Секвенирование геномов — это не только самые современные приборы, но и большой объем вычислений на суперкомпьютерах!
-
Авторы
-
Редакторы
Последний год жизни авторы этой статьи посвятили созданию инфраструктуры по получению, хранению и анализу кода жизни — генетической информации, которая записана в молекуле ДНК. Что такое ДНК с точки зрения математика, каковы основные принципы построения компьютерной архитектуры для анализа огромных массивов генетической информации и что ждать в будущем от тотальной прозрачности и доступности теперь уже и нашего индивидуального кода жизни, — обо всём этом расскажет предлагаемая вашему вниманию статья.
В своей знаменитой книге «Что такое жизнь с точки зрения физики?» Э. Шрёдингер описывал живую клетку с происходящими в ней процессами как динамическую систему, находящуюся в стационарном состоянии при неизменности внешних факторов. За прошедшие 65 лет с момента написания этой книги изменилось само представление о клетке и о том, как она реагирует на быстро меняющиеся условия обитания. На передний план вышли уже не те вещества, которыми клетка обменивается с окружающей средой в состоянии динамического равновесия, а информационные потоки, определяющие её развитие, рост и смерть в существенно неравновесных условиях. На смену биохимии пришли молекулярная биология и биоинформатика. Мы вплотную подошли к следующему эпохальному труду под названием «Что такое жизнь с точки зрения математики?», который терпеливо ждёт своего талантливого автора.
Сегодня все знают, что информация, необходимая оплодотворённой яйцеклетке, чтобы развиться сначала в эмбрион, а потом и во взрослый организм, записана в молекулах ДНК, последовательность нуклеотидных остатков в которой можно представить в виде текста. В этом тексте, как и в любом другом, самое важное — это последовательность букв (в ДНК их, как известно, всего четыре). Совокупность всех молекул ДНК ядра клетки (каждая из которых, взаимодействуя с белками, образует отдельные хромосомы) называют ядерным генóмом (митохондрии, бывшие в незапамятные времена свободноживущими микроорганизамами, имеют свой собственный геном). К примеру, бактерии, обитающие у нас в кишечнике и помогающие переваривать пищу, имеют геном длиной порядка нескольких миллионов букв. Простая вошь — уже 500 миллионов, а геном человека составляет более трех миллиардов букв. Для сравнения, все четыре тома «Войны и мира» Толстого содержат около двух миллионов букв, — т. е., примерно эквивалентны генетической информации бактерии, а геном человека можно сравнить со всей библиотекой Толстого в Ясной Поляне. (С другой стороны, объём кинофильма в формате Blu-ray уже существенно превосходит размер генома, — так что говорить нужно, конечно, не только об объёме информации, но и о её «качестве».)
В первом приближении можно считать, что все клетки взрослого человека имеют один и тот же генетический текст. («Нормальное» исключение составляют половые клетки и лимфоциты, а патологическое — клетки раковой опухоли.) Так же, как и в книге, где фрагменты текста объединяются в главы, в геноме протяжённые последовательности нуклеотидов объединяются в гены, контролирующие функции клетки и организма [1]. Лишь десять лет назад геном человека был в общих чертах «прочитан» — в 2000 году две команды исследователей объявили о независимом друг от друга завершении проекта по секвенированию (определению последовательности ДНК) генома человека. Результат их работы сейчас считается «золотым стандартом» генома человека (то, с чем исследователи, как с эталоном, сравнивают вновь полученные генетические тексты). Общие затраты на эту работу оцениваются в интервале 3–10 миллиардов долларов [2].
Читать — не перечитать
За прошедшие 10 лет технологии расшифровки генетических последовательностей развивались очень бурно, — в первую очередь, благодаря миниатюризации и автоматизации [3]. Следующее поколение методов секвенирования «информационных» молекул, будет, несомненно, связано с переходом к определению последовательности единичных молекул ДНК/РНК с применением нанотехнологических подходов. (Подробнее об этих технологиях, уже делающих первые уверенные шаги, «биомолекула» как-нибудь обязательно расскажет.)
Современная геномная лаборатория — например, Лаборатория геномики в Курчатовском Институте в Москве, — способна за один день «начитать» последовательность длиной до 20 миллиардов нуклеотидов. Что же касается стоимости таких работ, — прочтение одного человеческого генома уже подешевело почти до 10 000 долларов, — т. е., за 10 лет цена упала на 6 порядков (в миллион раз!). По оценкам экспертов, ценовой рубеж, когда персональная геномика войдёт в жизнь каждого из нас через медицину, страховки, работодателей и через прочие социальные институты, составляет 1000 долларов за индивидуальный геном. По всей видимости, он будет достигнут в ближайшие 5 лет.
Таким образом, теоретически можно ожидать, что через несколько лет каждый из нас будет обладателем 5 Гб информации о себе (именно столько занимают 3 миллиарда нуклеотидов в «четырехзначном исчислении», плюс служебные данные). Как хранить, анализировать и защищать эту важнейшую персональную информацию? Что она значит для жизни человека XXI века? Какие возможности и риски следует рассматривать в контексте обладания 5 Гб информации каждым из 6 миллиардов жителей Земли ?
Видимо, следует сделать оговорку — каждым из 6 миллиардов, достаточно обеспеченным, чтобы позволить себе тратить деньги на такие «пустяки». — Ред.
Внешние отличия, умственные способности и даже психологические особенности каждого человека в той или иной степени заложены в его геноме [4]. (Конечно, не стоит считать эти качества 100%-предопределёнными.) Считается, что основные генетические отличия одного человека от другого сосредоточены в однобуквенных заменах — своеобразных «опечатках» или «вариантах» текста ДНК, называемых однонуклеотидными полиморфизмами (ОНП). Как одна буква в названии книги «Война и мир» способна изменить его смысл (к примеру «Война и мор»), так и замены в генетических текстах могут привести к тому, что, например, одни люди будут болеть чаще, чем другие. Одни из нас — высокие и черноглазые, а другие — низкие и с голубыми глазами, и этим мы тоже обязаны генам. (И не только мы — например, «белая» окраска лошадей тоже обусловлена генетически [5].)
Зная генотип человека, теоретически можно предсказать многие его характерные черты, — не только цвет глаз и рост, но и предрасположенность к заболеваниям (именно это больше всего и интересует учёных и врачей) и даже к вредным привычкам [6]! Однако самое сложное здесь то, что большинство таких признаков определяется совокупностью большого, хотя и конечного числа «опечаток» в геноме, которые потребуется обнаружить. Результат появления той или иной «опечатки» не всегда бывает предсказуемым и понятным. Зачастую эффект от замены одной буквы слишком незначителен, и у исследователей нет ясности, как она в принципе может влиять на фенотип. Однако совокупность сотен и тысяч ОНП может эмпирически коррелировать с тем или иным признаком, хотя механизм, обусловливающий связь с признаком, может оставаться загадкой. Для этого необходимы данные о генотипах различных групп людей — как здоровых, так и больных, — чтобы иметь достаточную статистику для анализа отличий в геномах и поиска тех «опечаток», которые ответственны за склонность к заболеванию. (Подробнее о таких исследованиях, получивших название GWAS — genome-wide association studies — см. в статье «Загадочная генетика „загадочной болезни кожи“ — витилиго» [7].) Например, для проекта по исследованию рака почки генотипировано больше 12 тысяч человек. Таких проектов в мире можно насчитать уже несколько десятков, — то есть, всего насчитывается уже несколько сотен тысяч людей с установленными генотипами. Кстати говоря, уже и число полных геномов перевалило за тысячу [8].
Технология
Современные технологические платформы, предназначенные для чтения генетических текстов (для простоты такие приборы называют секвенаторами «нового поколения»), определяют последовательность не всей молекулы ДНК за раз, а лишь достаточно скромных её фрагментов — при большей длине, увы, возникает слишком много ошибок. Стадии секвенирования предшествует случайное «измельчение» ДНК на отрезки со средней длиной 500 нуклеотидов; само «считывание» осуществляется сразу с двух физических концов этого отрезка, в результате чего образуется пара фрагментов «текста» ДНК. Длина таких фрагментов, являющихся основным результатом работы секвенатора и на жаргоне именуемых «ридами» (от англ. read — читать), колеблется (в зависимости от производителя оборудования) в диапазоне 35–400 букв. Следует отметить, что большáя часть последовательности фрагмента неизбежно остаётся непрочитанной, поскольку длины «рида» (допустим, 100 букв) не хватает, чтобы перекрыть всю длину фрагмента (≈500 букв).
Подробнее об инструментальных основах секвенирования мы уже писали (см. «454-секвенирование (высокопроизводительное пиросеквенирование ДНК)» [3]); сейчас же разговор пойдёт главным образом о том, как скомбинировать из огромной библиотеки фрагментов текста «готовую» генетическую последовательность, как она записана в геноме. Современный прибор за один запуск длительностью около 10 дней способен сгенерировать до 300 миллионов пар «ридов» (рис. 1). Точная сборка этого «стройматериала» в геномную последовательность — проблема не из лёгких, и решаться она может двумя путями, в зависимости от наличия «эталонного» генома.

Рисунок 1. Секвенаторы SOLiD компании Applied Biosystems (Life Technologies) в лаборатории Queensland Centre for Medical Genomics, Австралия. Одна из миссий центра — «разрабатывать и улучшать технологию для рутинного анализа полной последовательности человеческого генома». Научных институтов, ориентированных на развитие «персонализованной медицины», с каждым годом становиться все больше и больше.
Задача первая: ресеквенирование
Возвращаясь к аналогии с произведением Льва Николаевича, ресеквенирование можно сравнить с поиском опечаток в исходном тексте «Войны и мира». Мы считаем, что геном изучаемого объекта имеет то же строение, что и «золотой стандарт» генома 2000-го года (хотя и этот эталон постоянно уточняется — сейчас доступна уже 37-я его версия). Сопоставляя участки текста, для каждого из многих миллионов «ридов» определяются его координаты на одной из хромосом: «страница», «абзац», «отступ слева» и т. д. В случае если обнаруживается расхождение с эталоном — опечатка, маленькая вставка или выпадение текста (на жаргоне такие отличия называют short indels — short insertions/deletions), — эти вариации включаются в отчёт о сравнении. Так, сравнивая миллионы и миллиарды «ридов» с исходным текстом, можно получить полный перечень отличий изучаемого генома от эталонного «золотого стандарта». Более того, если каждая буква исходного текста проверяется многократными прочтениями, это увеличивает статистическую достоверность найденных генетических особенностей и аномалий. Сегодня считается, что геном ресеквенирован с высоким «покрытием» (deep sequencing), если каждая его буква была прочитана в среднем 30 раз или более (30×).
С точки зрения биоинформатических алгоритмов, ресеквенирование — это относительно лёгкая процедура: для обработки данных от одного запуска прибора требуется всего около 10 часов работы программы на 20 процессорных ядрах и 20 Гб оперативной памяти.
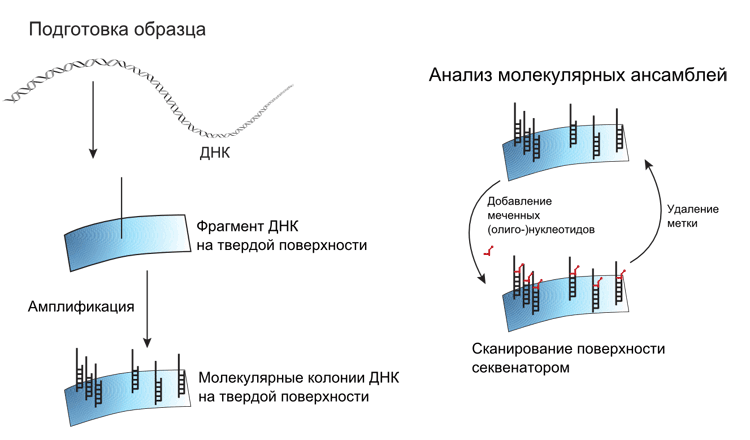
Рисунок 2. Общий принцип секвенирования нового («второго») поколения. Первый этап процесса подготовки образца состоит из фрагментации длинных молекул ДНК ультразвуком или каким-нибудь другим методом, не зависящим от последовательности ДНК, до размера 250–500 пар нуклеотидов. Далее следует стадия, во время которой концы ДНК-фрагментов приводятся в порядок: сначала удаляются или достраиваются выступающие концы молекул, затем лигируются адаптерные олигонуклеотиды. Полученная библиотека предварительно амплифицируется для увеличения представленности каждого фрагмента ДНК. Последний шаг — создание молекулярных колоний — клональная амплификация каждого фрагмента библиотеки на твердой поверхности. Все это происходит вне секвенатора. Расшифровка последовательности ДНК-фрагментов происходит через параллельный анализ миллионов и даже миллиардов молекулярных ансамблей в секвенаторе. В приборе стадии подачи реактивов для секвенирования (нуклеотидов или олигонуклеотидов с флуорофором и соответствующих ферментов) чередуются со стадиями сканирования поверхности и регистрации получаемых изображений.
Задача вторая: секвенирование de novo
Вторая задача является и экспериментально, и алгоритмически, и вычислительно более сложной — тут требуется реконструировать текст из набора «ридов», не имея эталона для сборки. Подход основан на том, что, в силу случайности разбиения молекул ДНК на фрагменты, при достаточно плотном «покрытии» обязательно найдутся несколько частично перекрывающихся «ридов», при совмещении которых текст будет постепенно наращиваться. «Подрастающий» текст (в данном случае называемый контигом) используется для поиска среди миллиарда «ридов» такого, который максимально (но не полностью) с ним перекрывается.
Процедура объединения контигов продолжается до тех пор, пока с обоих концов фрагмента генетического текста не начнутся области протяжённых повторов, характерные для «кончиков» хромосом. Если повтор имеет длину большую, чем длина «рида», то его длина, а значит, и точная последовательность, остаётся неизвестной. Однако здесь на помощь приходит информация о парности чтений: как правило, они находятся на более-менее известном расстоянии друг от друга. Таким образом, если одно из чтений пары попадает на один контиг, а второе — на другой, то эти контиги можно объединить в связку, называемую скаффолдом. Впоследствии непрочитанные «дыры» в скаффолдах можно будет прочесть другими методами. Сборка de novo является алгоритмически сложным и вычислительно затратным процессом.

Рисунок 3. Повторное секвенирование («ре-секвенирование») генома с целью выявления разнообразных структурных вариаций (однонуклеотидных полиморфизмов, или «снипов», а также инсерций, делеций, повторов, инверсий, транслокаций). В отличие от секвенирования неизвестных последовательностей de novo, при котором прочтения соотносятся друг с другом и собираются в контиги, для ре-секвенирования достаточно просто «картировать» прочтения на референсную последовательность, уже имеющуюся под рукой. Снипы выглядят как однонуклеотидные замены в коротких прочтениях, при этом количество прочтений с заменой говорит о состоянии аллеля — гомозиготном (все прочтения с заменой) или гетерозиготном (половина прочтений с заменой).
Такие задачи решаются с использованием теории графов, но идеального «сборщика» текстов для de novo-секвенирования ещё не создали. Основной проблемой сборки является наличие в генетических текстах длинных (от 200 до нескольких тысяч букв) элементов, содержащих повторы длиной от 4 до 150 нуклеотидных оснований. Очевидно, что именно из-за присутствия повторов текст во время сборки может оборваться. Для преодоления этого используют экспериментальные ухищрения, заключающиеся в генерации исходной библиотеки фрагментов со средней длиной не 500, а 3000 или даже 10000 букв. В этом случае существенно увеличивается вероятность захватить парой «ридов» уникальные участки текста, оставив повторы внутри.
Описав очень схематично схему алгоритма секвенирования, приступим к описанию вычислительных мощностей, которые авторы привлекают для анализа генетических текстов.
Секвенирование: без суперкомпьютеров не обошлось
«Сборка» текста генома из набора фрагментов, полученных на секвенаторе, — алгоритмически и вычислительно сложная задача, невозможная без использования суперкомпьютерных кластеров. Например, каждая сборка генома печёночного сосальщика, основанная на данных нескольких запусков секвенатора, требует до недели работы кластера из двух десятков узлов по 8 ядер и 8 Гб оперативной памяти в каждом (объединение по интерфейсу MPI). Однако одного запуска почти всегда недостаточно — таких сборок может быть несколько из-за необходимости подбора оптимальных параметров алгоритма и добавления новых экспериментальных данных. Есть и альтернативные варианты решения этой задачи, основанные не только на кластерах, но и популярных сегодня «облачных» вычислениях.
В целом, сборка de novo является более перспективным методом, чем ресеквенирование, и практически единственным подходом — когда эталонной последовательности генома исследуемого организма ещё не существует (как правило, для этого и проводится первое секвенирование). Оно позволяет выявлять существенные перестройки в геноме, обозревая его как одно целое. Впрочем, для многих практических целей и ресеквенирования бывает вполне достаточно.
Запрограммируй это
Читателю уже должно было стать ясно, что без серьёзных компьютерных мощностей решить задачу секвенирования генома шансов немного. Некоторые исследователи видят решение проблемы доступности дешёвых вычислений в так называемых «персональных суперкомпьютерах», под которыми имеются в виду системы на базе графических процессоров. Действительно, их специализация на операциях с векторами и массивная параллелизация находят всё более широкое применение во многих областях науки. В то же время относительно низкая цена и стоимость и владения такими компьютерами существенно снижают порог вхождения; их могут позволить себе не только институты, но и отдельные лаборатории.
Однако переход на новую технологию обязательно приносит с собой ряд проблем, и в биоинформатике они ощущаются особенно остро. В частности, часто необходимо использовать специальные алгоритмы, требующие от программиста знания архитектуры графических процессоров; в то же время большинство программного обеспечения в биологии разрабатывается биологами, для которых программирование — лишь дополнительный навык. Это же обусловливает и приверженность биологов к скриптовым языкам программирования: часто требуется написать простую программу «на раз» — только для проверки очередной гипотезы. Традиционно используемый в биоинформатике язык Perl на настоящий момент не имеет доступа к OpenCL (программный комплекс для облегчения программирования графических процессоров), хотя некоторые другие языки, например, Python или Java, уже оснащены привязкой к этому фреймворку.
Можно назвать три основных особенности использования графических сопроцессоров в геномной биоинформатике. Во-первых, это текстовый формат геномных данных, в то время как единого стандарта для их представления в удобном (с точки зрения вычислений, компактном и быстром) цифровом виде до сих пор нет. Во-вторых, высокая «квантуемость» данных («ридов» секвенатора, полиморфизмов и др.) способствует многопоточной обработке. И, в-третьих, огромные требования к оперативной памяти, запас которой непосредственно на графическом ускорителе пока относительно мал; это может привести к дополнительному усложнению и без того порой неочевидных алгоритмов.
Требования к аппаратному обеспечению, накладываемые практическими задачами, почти всегда опережают реальные возможности вычислительных машин. Для многих приложений биоинформатики — таких как сборка геномов de novo — часто не хватает ресурсов даже самых современных кластеров, включающих сотни вычислительных узлов, соединенных быстрой сетью Infiniband.
Возможное решение проблемы компьютерных мощностей — глобальная грид-инфраструктура, объединяющая десятки суперкомпьютерных центров и позволяющая использовать их мощности через единый интерфейс. Кроме того, грид-технология позволяет создавать распределённое хранилище данных, — а ведь когда речь идёт о геномике, дискового пространства не бывает много. Последнее время это направление распределённых вычислений очень активно развивается, — например европейский проект EGEE является примером создания крупнейшей грид-инфраструктуры, объединяющей участников из более чем 50 стран и включающей в себя более 260 компьютерных центров. Общее количество вычислительных ядер в этой сети более 150 тысяч, а дисковое пространство превышает 28 петабайт. Возможно, использование грид-технологий сможет отодвинуть границу доступных задач в биоинформатике уже в самом ближайшем будущем.
Заключение
Как же будет выглядеть персональная геномика через несколько лет? Нам видится, что буквально в каждом крупном городе будет лаборатория по чтению индивидуальных геномов. Скорее всего, не стоит опасаться того, что индивидуальная информация станет доступна лаборанту, обслуживающему секвенатор — он все равно не сможет её интерпретировать. Хотя, безусловно, сохранности персональной тайны пациента следует уделить самое пристальное внимание. Последовательности геномов будут поступать в единый информационный центр, который проведёт автоматизированный анализ и сравнение с геномами других людей, для которых уже создана история болезни. На основании этого можно будет делать индивидуальный прогноз для каждого пациента, который, быть может, ещё и не пациент вовсе, а всего лишь новорожденный.
Более того, все болезни, физические параметры, психофизиологические особенности, наблюдаемые у него в будущем, также можно будет занести в его персональный файл и соотнести с генетическим текстом. Приходя на приём к врачу, человек получит рекомендации по лечению и приёму лекарств, наиболее соответствующих его метаболизму и предрасположенностям.
| Название учреждения | Платформа |
|---|---|
| РНЦ «Курчатовский институт», Москва | Illumina GA IIx (×3) Applied Biosystems SOLiD 4 (×2) |
| НИИ химической биологии и фундаментальной медицины СО РАН, Новосибирск | Applied Biosystems SOLiD 4 454/Roche FLX Titanium |
| Инновационно-технологический центр «Биологически активные соединения и их применение» РАН, Москва | llumina GA IIx |
| Институт общей генетики им Н. И. Вавилова, Москва | Applied Biosystems SOLiD 4 llumina HiSeq 2000 |
| Институт молекулярной биологии им. В. А. Энгельгардта РАН, Москва | llumina GA IIx |
| Генаналитика, Москва | Applied Biosystems SOLiD 4 |
| Секретная станция переливания крови, Киров | 454/Roche FLX Titanium |
| Лимнологический институт СО РАН, Иркутск | 454/Roche FLX Titanium |
| Центр «Биоинженерия» РАН, Москва | 454/Roche FLX Titanium |
| НИИ физико-химической медицины, Москва | Applied Biosystems SOLiD 4 |
| Биологический факультет МГУ им. М. В. Ломоносова, Москва | Applied Biosystems SOLiD 4 |
...Над всем этим неумолимо нависает призрак киберпанка, предвосхищая растворение человека в океане цифр и знаков. Большой брат будет следить и за тобой. Но только если ты этого захочешь. Это наступит неотвратимо, но не через один год жизни...
Первоначально сокращённый вариант этой статьи был опубликован в журнале «Суперкомпьютеры» [9].
Статья написана в соавторстве с Чекановым Н.Н. и Теслюком А.Б. при участии коллектива «биомолекулы». Врезку, таблицу, картинки и «словарик» подготовил Павел Натальин.
Литература
- Krebs J.E., Goldstein E.S., Kilpatrick S.T. Lewin's GENES XII (12th Edition). Jones & Bartlett Learning, 2017. — 838 p.;
- Геном человека: как это было и как это будет;
- 454-секвенирование (высокопроизводительное пиросеквенирование ДНК);
- Слово о генетике поведения;
- Код белой лошади;
- Спасибо, дорогой Минздрав, что предупредил!;
- Загадочная генетика «загадочной болезни кожи» — витилиго;
- Перевалило за тысячу: третья фаза геномики человека;
- Прохорчук Е.Б. (2010). Код жизни. Суперкомпьютеры. 2, 38–41;
- Скрябин К.Г., Прохорчук Е.Б., Мазур А.М., Булыгина Е.С., Цыганкова С.В., Недолужко А.В. и др. (2009). Комбинирование двух технологических платформ для полногеномного секвенирования человека. Acta Naturae. 3, 113–119;
- Grigorenko A.P., Borinskaya S.A., Yankovsky N.K., Rogaev E.I. (2009). Achievements and Peculiarities in Studies of Ancient DNA and DNA from Complicated Forensic Specimens. Acta Naturae. 3, 64–76;
- Carl W Fuller, Lyle R Middendorf, Steven A Benner, George M Church, Timothy Harris, et. al.. (2009). The challenges of sequencing by synthesis. Nat Biotechnol. 27, 1013-1023.

Комментарии


