Теоретическая химия: от молекулы водорода до структуры белков
03 ноября 2016
Теоретическая химия: от молекулы водорода до структуры белков
- 2941
- 0
- 10
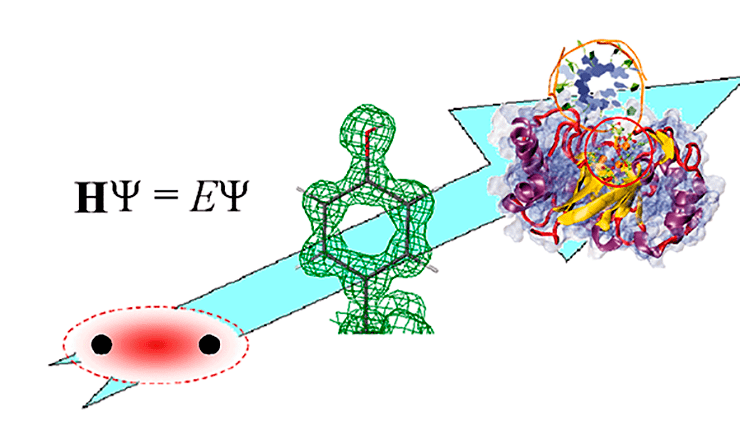
От понимания природы химической связи до моделирования работы белковых комплексов и клеточных структур — такой путь прошла теоретическая химия всего за 100 лет
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: Сто лет назад Гилберт Н. Льюис рисовал химические формулы с точками, обозначающими электроны, а Нильс Бор формулировал постулаты квантовой теории и объяснял строение атома. Эта статья о том, как эволюционировали представления ученых о химической связи, как эти представления помогли увидеть структуру молекул, а знания о молекулярной структуре помогли развитию теории, и как ученые пришли к искусству моделирования живых белков в действии.

«Био/мол/текст»-2016
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2016.
Генеральным спонсором конкурса, согласно нашему краудфандингу, стал предприниматель Константин Синюшин, за что ему огромный человеческий респект!
Спонсором приза зрительских симпатий выступила фирма «Атлас».
Спонсор публикации этой статьи — Артур Залевский.
Философские атомы
То, что все тела состоят из атомов, ученые подозревали еще в античности. «Атом» по-гречески означает «неделимый». В XVIII веке появились первые косвенные доказательства того, что все тела не являются сплошными и непрерывными, но состоят из мельчайших частиц, дальше которых дробить вещество невозможно. Более того, было обнаружено, что частицы эти могут соединяться друг с другом не как попало, а только в определенных пропорциях и в определенном порядке.
Однако до конца XIX века атом был скорее умозрительным, абстрактным понятием. Большинство химиков пользовалось им вынужденно, например, когда говорили, что «частица» воды содержит один атом кислорода и два атома водорода или что атомы соединяются в частицу в определенном порядке и влияют друг на друга. Слово «атом» в этом контексте означало не физическую частицу, а некую порцию вещества, реагирующую с другими веществами в определенном соотношении. Из тех же времен происходит термин «эквивалент», означающий количество вещества, реагирующего в точности без остатка с заданным количеством другого вещества в определенной реакции. Понятно, что для разных реакций эквиваленты одного и того же вещества были разными, что создавало изрядную путаницу. Были и «мастодонты» типа Вильгельма Оствальда (Нобелевская премия по химии 1909 г.), не признававшие концепции атомов вообще .
Оствальд умудрялся обходиться в своих сочинениях без атомов в смысле частиц, и следы этого неприятия мы находим в данном им и принятым международным сообществом определении единицы количества вещества — моля: «Моль есть количество вещества системы, содержащей столько же структурных элементов, сколько содержится атомов в углероде-12 массой 0,012 кг». Более современное, но пока еще не принятое Международным бюро мер и весов, определение моля базируется на фиксированном значении постоянной Авогадро (моль — это 6,022140857·1023 частиц) и не зависит от определения единицы массы. А Оствальд смог изменить свое отношение к атомистической теории после опытов Жана Перрена по броуновскому движению.

Рисунок 1. Шаростержневая модель метана (обратите внимание, он здесь плоский), которой пользовался на лекциях Август Вильгельм Гофманн.
сайт wikimedia.org
Тем временем химикам-органикам часто приходилось сталкиваться с веществами, имеющими одинаковый состав, но совершенно разные свойства — изомерами. Когда в головах ученых устоялась теория, гласящая, что свойства вещества зависят от того, в каком порядке соединились его атомы, умозрительным атомам пришлось приписать форму. В частности, атом углерода стали считать тетраэдрическим [1]. Тогда же появились первые шаростержневые модели молекул, такие как на рисунке 1.
Но что (кроме палочек или крючочков на деревянных модельках) заставляет атомы соединяться друг с другом? В 1898 г. Людвиг Больцман выдвинул идею об особых «чувствительных областях» на поверхности атома. Атомы образуют молекулу, только если они контактируют друг с другом этими чувствительными областями — тогда между ними возникает притяжение, чувствительные области перекрываются (sic!) и образуется химическая связь (рис. 2).

Рисунок 2. Диаграмма Людвига Больцмана, показывающая перекрывание «чувствительных областей» α и β в молекуле I2.
сайт wikimedia.org
Атомы становятся реальными
Тем временем к началу XX века были открыты положительные ионы с разным соотношением заряда и массы (1886 г., Ойген Гольдштейн) и частица, несущая элементарный отрицательный заряд, — электрон . Жан Батист Перрен в 1908 г. доказал существование молекул [2]. И, наконец, в 1909 г. Эрнест Резерфорд провел свой знаменитый эксперимент с бомбардировкой золотой фольги α-частицами. В этом эксперименте он установил, что атом состоит из маленького положительно заряженного ядра, вокруг которого летают электроны. Так что для физиков атомы были вовсе не фикцией.
Я намеренно не называю имени «первооткрывателя» электрона, т.к. его обнаружили и идентифицировали по соотношению заряда и массы в 1896 г. и в катодных лучах (Джозеф Томсон, Джон Таунсенд, Г.А. Уилсон), и в β-излучении радиоактивных материалов (Анри Беккерель), а Роберт Милликен с Харви Флетчером в 1909 г. и — независимо от них — Абрам Фёдорович Иоффе в 1911 г. измерили его абсолютный заряд.
Одним из первых, кто заподозрил, что в деле химической связи замешаны электроны, был Гилберт Ньютон Льюис. Был он в большей степени физиком, чем химиком, занимался термодинамикой, интересовался только-только возникшей квантовой теорией, так что про электроны был в курсе. Кандидатская диссертация Льюиса была посвящена электрохимическим потенциалам, и впоследствии он стажировался у В. Нернста в области электрохимии. Квантовая физика в сочетании с электрохимией дала интересный эффект.
В 1904 г. Ричард Абегг, также бывший в 1894 г. сотрудником у Нернста, разрабатывал т.н. электрохимическую теорию валентности. В частности, он обнаружил, что разность между высшей степенью окисления элемента (например, в его оксиде) и его низшей степенью окисления (в гидриде) часто равна 8. Если вспомнить, что степень окисления в оксиде — это количество электронов, которое атом может отдать, а степень окисления в гидриде — сколько он может принять, то валентность оказывается завязанной на магическое число 8: либо атом отдает лишние электроны, оставаясь с 8 электронами «на борту», либо принимает недостающие до 8. Именно это и стало ключевой идеей для Льюиса, превратившись в знакомое нам со школы «правило октета»: ионы или атомы с заполненной оболочкой из 8 электронов обладают особой стабильностью.
Читая лекции студентам Гарварда, Льюис пытался объяснить им понятие валентности, изображая атомы в виде кубиков, в вершинах которых сидят электроны: 8 вершин, частично заполненных электронами (рис. 3). Атомы второго периода он представлял однослойными кубиками, третьего — двухслойными (таким образом он ввел понятие электронных оболочек). Были в истории химии шарики с крючочками, были тетраэдры, теперь вот до кубиков договорились... На что только не пойдешь, чтобы втолковать материал студентам!

Рисунок 3. Рисунок Льюиса из материалов лекций 1902 года.
сайт wikimedia.org
Связи между атомами образуются путем обобществления электронов так, чтобы на каждом атоме их получалось 8. На кубиках это выглядело так — рисунок 4.

Рисунок 4. Связи между атомами. а — Несвязанные атомы. б — Промежуточное состояние с одним обобществленным электроном. в — Одинарная связь, образованная обобществленной парой электронов. г — Двойная связь, образованная двумя обобществленными парами электронов. Тройную связь изобразить уже не удается.
В 1913 году вышла статья Альфреда Парсона, где он предположил, что электрон — это миниатюрный магнит, и химическая связь образуется в результате притяжения этих магнитиков. Паззл сощелкнулся! В 1916 г. выходит статья Льюиса «The Atom and the Molecule» [3], где химические связи обозначаются палочками, а электроны — точками (знакомые нам со школы льюисовские структуры), например, так — рисунок 5.

Рисунок 5. Примеры льюисовских структур.
Впрочем, модель кубического атома Льюиса прожила даже меньше, чем модель «булки с изюмом» Джозефа Томсона, где изюминки-электроны были напиханы в положительно заряженную булку. И та, и другая модель были выдуманы из головы. «Булочная» модель не выдержала экспериментальной проверки в опытах Резерфорда. А кубические атомы пали под натиском теории: в 1913 г. Нильс Бор предложил модель атома, основанную на стационарных орбитах электронов. Из трех простых постулатов (т.н. постулаты Бора) и законов сохранения следовала необходимость квантования энергии и углового момента электрона, а из этого квантования — правила заполнения электронных оболочек в том виде, в котором мы их знаем со школы [4]: на 1-й оболочке два электрона, на 2-й — восемь (вот он, октет!), на 3-й — уже 18. С кубиками, вложенными один в другой, эта модель никак не согласовывалась. А в 1926 г. задачу об электроне в атоме решили на основе волновой механики Эрвина Шрёдингера, не привлекая никаких постулатов (т.е. из первых принципов), и получили картинку, в точности соответствующую боровскому атому.
В 1928 г. Поль Дирак включил в задачу релятивистские эффекты, связанные с высокой скоростью движения электрона вокруг массивного ядра, и пришел к идее спина электрона. Таким образом, даже идея Парсона о магнетизме электрона нашла подтверждение, хотя магнетизм этот оказался несколько необычным. После этого модель строения атома приобрела свой современный вид.
Если физики не сомневались в существовании атомов, то остальное научное сообщество еще требовалось в этом убедить. Первые эксперименты по рентгеновской дифракции были проведены в 1912 г. Максом фон Лауэ, в результате которых была определена кристаллическая структура сульфата меди — элементарная ячейка и положение атомов в ней. За эту работу фон Лауэ получил Нобелевскую премию по физике в 1914 г. После этого последовали расшифровки огромного количества кристаллических структур неорганических и органических веществ.
Однако наиболее прямую демонстрацию атомов удалось получить уже после Второй мировой войны — в 1951 г., когда Эрвин Мюллер изобрел ионный проектор (разновидность электронного микроскопа) и с его помощью получил изображение атомов платины на острие иглы (рис. 7). С тех пор изобрели много разных конструкций электронного микроскопа, позволяющих ощупывать и визуализировать поверхности материалов на атомном уровне. А в 2013 г. IBM сняли мультфильм «A Boy and His Atom», нарисованный молекулами окиси углерода на медной подложке с помощью сканирующего туннельного микроскопа (см. видео). Теперь мы можем буквально трогать атомы руками и манипулировать ими [5]!

Рисунок 7. Изображение кончика платиновой иглы в ионном проекторе. Яркие точки — атомы.
сайт wikimedia.org
Мультфильм «Мальчик и его атом», нарисованный молекулами окиси углерода на медной подложке
Но вернемся к статье Льюиса 1916 г. Она оказала большое влияние на юного студента Орегонского Кулинарного техникума Сельскохозяйственного колледжа Лайнуса Полинга, который в 1922 г. с дипломом химика-технолога отправился в Калтех изучать рентгеновскую кристаллографию, а в 1926 г., уже защитив диссертацию, получил грант на стажировку в Европе. Там он работал в Мюнхене у Арнольда Зоммерфельда, в Копенгагене у Нильса Бора и в Цюрихе у Эрвина Шрёдингера. Неплохую подготовку давали в американских сельхозтехникумах, не правда ли?
В дело вступают физики
В 1927 г. Эйвинд Буррау точно решил из первых принципов задачу об электроне в поле двух ядер, т.е. о молекулярном ионе H2+. Эта работа показала, что задачу о химической связи можно решать методами квантовой механики, хотя задействованные при этом математические приемы оказались неприменимыми в общем случае. И в том же году Вальтер Гайтлер и Фриц Лондон приближенно решили задачу о молекуле водорода H2, сконструировав волновую функцию молекулы (спасибо Шрёдингеру) из двух волновых функций отдельных атомов водорода в виде их суммы и разности, и таким образом показали, как образуется ковалентная связь. Качественный результат оказался очень похожим на решение задачи про H2+. И при этом Льюис оказался прав! Электроны в молекуле H2 действительно принадлежат обоим атомам в равной мере, хотя картинки, где два точки-электрона сидят промеж двух ядер, получиться не может в принципе: все-таки электрон обладает волновой природой и не может сидеть на одном месте. Вместо этого наблюдают увеличение плотности вероятности нахождения электрона на линии, соединяющей атомы (рис. 8).

Рисунок 8. Плотность вероятности обнаружения электрона в молекуле H2, вид сбоку и сверху (черные точки — ядра, красное — электронная плотность)
Немного позже, в 1929 г., задача о молекуле водорода была решена еще раз, другим методом. В 1926–1927 гг. Фридрих Хунд и в 1927–1928 гг. Роберт Малликен ввели понятие молекулярной орбитали — волновой функции одного электрона в поле ядер. А в 1929 г. Джон Леннард-Джонс предложил строить молекулярные орбитали в виде линейных комбинаций атомных орбиталей и решать уравнение Шрёдингера для них. Тогда для описания молекулы нужно взять все атомные орбитали и «просто» построить из них нужные линейные комбинации. При таком описании все электроны в молекуле в той или иной степени принадлежат всем атомам. Этот подход получил название теории молекулярных орбиталей.
Тем временем, идея Гайтлера и Лондона тоже оказалась плодотворной, ведь таким образом можно описывать любые ковалентные связи, а целую молекулу — как совокупность ковалентных связей между соседними атомами. Нужно «просто» взять волновые функции электрона на одном атоме связи и на другом атоме и построить их комбинацию. Метод получил название теории валентных связей. Вот только у атома углерода, например, есть два s-электрона и два p-электрона, какой взять, чтобы образовать связь C—H в метане, если вспомнить, что все C—H связи в метане эквивалентны и направлены по углам тетраэдра? Лайнус Полинг в 1931 г. предположил, что в присутствии атомов-партнеров (в данном случае, водорода) орбитали углерода гибридизуются и превращаются из сферической s и трех гантелеобразных p в вытянутые фиговины, направленные по углам тетраэдра. Эти гибридные орбитали имеют вид (s + √3p), где за p-орбиталь берется та, что направлена вдоль соответствующей C—H связи. Из четырех исходных орбиталей получаются четыре новые, т.н. sp3-гибридные, и на каждой сидит по одному электрону, готовому образовать связь с атомом водорода (рис. 9).

Рисунок 9. Перекрывание sp3-гибридных орбиталей углерода (серые) и атомных s-орбиталей водорода (синие).
В том же 1927 г. Дуглас Хартри предложил для решения многоэлектронного уравнения Шрёдингера метод самосогласованного поля. В этом методе предполагается, что каждая частица двигается в некотором усреднённом самосогласованном поле, создаваемом всеми остальными частицами системы, и таким образом многочастичную задачу сводят к одночастичной. В 1930 г. Джон Слейтер и Владимир Александрович Фок улучшили этот метод с учетом принципа Паули (антисимметрии волновой функции по отношению к перестановке пары электронов) и вариационного принципа. Метод получил название метода Хартри—Фока. В современном (с 1951 г.) варианте он используется в комбинации с методом молекулярных орбиталей.
С появлением методов МО и ВС у квантовых химиков разыгрался аппетит. Хотелось научиться строить волновые функции не только для простейших молекул, но и для чего-то более сложного. Однако расчеты, основанные только на принципах квантовой механики и основных физических константах — т.н. расчеты из первых принципов — оказались исследователям не по зубам. Объем вычислений рос катастрофически с размером задачи. Поэтому для практических целей на основе методов МО и ВС разработали т.н. полуэмпирические методы, где часть параметров не вычислялась, а бралась из эксперимента. Тем не менее до появления компьютеров квантовохимические расчеты многоатомных молекул были редкостью.
А может, обойдемся без волновой функции?
Пока сторонники методов ВС и МО спорили, чье решение правильнее, в том же 1927 г. Люэлин Томас и Энрико Ферми независимо разработали квазиклассическую статистическую модель атома, базирующуюся на концепции электронной плотности. То есть вместо индивидуальных волновых функций N электронов, каждая из которых зависит от трех переменных (x, y, z), так что вся задача становится 3N-мерной, предлагалось работать с электронной плотностью — вероятностью обнаружить электрон в малой окрестности заданной точки. Электронная плотность уже возникала как побочный продукт в методах волновых функций (МО и ВС), но в методе функционала плотности электронная плотность, зависящая только от трех координат, стала главным элементом. Потеря информации о судьбе каждого отдельного электрона оказалась несущественной — информации о распределении электронной плотности оказалось достаточно, чтобы вывести все свойства молекулы.
Несмотря на всю привлекательность для вычислений, теория Томаса-Ферми-Дирака (последний внес в нее существенные дополнения) оставалась совершенно непригодной для описания молекул и ковалентно связанных кристаллов, но худо-бедно использовалась для расчетов металлов и полуметаллов. Впоследствии, в 1964 году, теория функционала плотности была переформулирована так, чтобы исключить из нее грубые приближения теории Томаса-Ферми. В таком виде теория функционала плотности дожила до разработки достаточно точных и универсальных функционалов и к настоящему времени стала едва ли не основным инструментом квантовых химиков.
Не молекулой единой
В 1928 г. (до чего же продуктивное было время для квантовой механики!) Феликс Блох решил задачу об электроне в кристалле. В отличие от свободного атома, волновые функции электрона в периодическом потенциале ядер оказались похожими на плоские волны. А в 1930 г. Леон Бриллюэн ввел в теорию твердого тела понятие энергетических зон. Зоны можно представить как предельный случай комбинации большого количества атомных орбиталей (рис. 10). В пределах одной зоны энергия электронов изменяется непрерывно, а не дискретно, как на орбиталях молекулы. Между зонами есть промежуток — запрещенная зона, — в которой энергетических уровней нет. Ее электроны могут преодолеть только скачком, как в молекулах, так и в атомах. Свободное перемещение электронов внутри зоны обеспечивает металлическую проводимость, а благодаря относительно небольшой ширине запрещенной зоны становится возможной проводимость полупроводников. Если же запрещенная зона достаточно широкая, получится диэлектрик или изолятор.

Рисунок 10. Образование зон как предельный случай комбинации бесконечного числа атомных орбиталей.
сайт lugovsa.net
Так получилось, что наука о материалах первое время имела дело с кристаллическими материалами, и аппарат зонной теории был там весьма уместен. А в сочетании с теорией функционала плотности зонная теория позволяет надежно прогнозировать свойства материалов.
Провал во времени
Да, 1927–1930 гг. были исключительно щедрыми на основополагающие открытия в области квантовой химии. А потом — как отрезало. Что случилось? Случился приход к власти Гитлера в 1933 г. Большинство физиков-теоретиков жили в Германии и соседних странах. Почему-то так случилось, что они оказались крайне неугодны фашистскому режиму: кто из-за своего еврейского происхождения, а кто — из-за сочувствия к коллегам-евреям. Начался массовый исход физиков сначала из Германии, а затем и из соседних стран: Голландии, Швейцарии и т.д. Уезжали, в основном, в Америку, а там ученым-эмигрантам приходилось заниматься не тем, к чему душа лежит, а тем, на что есть заказ — в основном, прикладными проблемами. Кто-то попал в Манхэттенский проект, кто-то полностью перешел на преподавание. Тем, кто уехал в Америку, повезло.
Ганс Гельман был социалистом по убеждениям и уехал от фашистов в Советский Союз. С 1933 по 1938 г. он работал в Физико-химическом институте Карпова в Москве, где написал первый в мире учебник по квантовой химии, а в 1938 г. был арестован, обвинен в шпионаже в пользу Германии и расстрелян.
Те физики, что никуда не эмигрировали, тоже занимались в основном прикладными задачами и не отвлекались на баловство типа строения молекул. За исключением нашего знакомого Лайнуса Полинга.
Полинг вернулся в Америку после стажировок в Европе. С 1927 г. он занимался рентгеновской кристаллографией и с 1930 г. электронографией — исследовал молекулярную структуру самых различных веществ, а параллельно занимается теорией химической связи. Серия статей «The nature of the chemical bond» выходит в 1931 г. [6–10], а книга «The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry» [11], названная «библией химии», вышла первым изданием в 1939 г. Помимо гибридизации, о которой рассказано выше, он ввел понятие электроотрицательности и построил шкалу электроотрицательности элементов. С помощью электроотрицательности можно судить о степени ионности (полярности) ковалентной связи. Еще одним достижением Полинга было объяснение строения ароматических углеводородов, в частности, бензола. Структура с чередующимися двойными и одинарными связями не согласовалась с наблюдаемыми химическими свойствами бензола. Полинг предположил, что структура реального бензола является суперпозицией двух структур (рис. 11), а не их быстрого взаимопревращения. Впоследствии он дал этому явлению название резонанса .
Почему-то концепция резонанса (суперпозиции структур) вызвала резкое неприятие у советских химиков, вылившееся в разгром квантовохимической науки в СССР в 1951 г. — «дело о теории резонанса». Кампания по борьбе с резонансом фактически носила ритуально-идеологический характер: достоинства и недостатки теории как таковой не обсуждались, разгрому подвергались лично сторонники теории, причем их доводы во внимание не принимались. В результате квантовая химия стала наукой, которой опасно заниматься: один из главных злодействующих лиц, проф. В.М. Татевский, до самой своей смерти в 1999 г. работал на химфаке МГУ и крайне бурно реагировал на упоминание молекулярных орбиталей.

Рисунок 11. Резонансные структуры бензола
В конце 1930-х Полинг увлекся биомолекулами. Так, он исследовал гемоглобин и показал, что его структура меняется при обратимом присоединении кислорода. Во время Второй мировой войны Полинг работал в частности и над биомедицинскими приложениями. А после войны занялся структурой белков всерьез. Начиная эту работу, он надеялся научиться теоретически предсказывать структуру белков [12]. Хммм... ну-ну...
Впрочем, в 1951 г., зная только структуры отдельных аминокислот и коротких пептидов и сделав предположение о плоской структуре пептидной связи, Полинг с сотрудниками пришел к выводу о том, что α-спираль и β-лист являются основными структурными мотивами во вторичной структуре белков [13]. А вот в гонке за расшифровкой структуры ДНК Полинг и его команда взяли мощный фальстарт: опубликовали статью, где предлагали для ДНК структуру тройной спирали с нейтральными фосфатными группами — совершенно неверную, противоречащую экспериментальным данным. Это, однако, не охладило их пыл, и в дальнейшем Полинг продолжал заниматься изучением структуры и функций белков, ферментными реакциями и связью болезней со свойствами отдельных белков и кодирующими их генами. В частности, в 1949 г. Полинг с сотрудниками показали, что серповидноклеточная анемия вызывается дефектным гемоглобином, который наследуется по Менделю. В 1954 г. Полинг получает заслуженную Нобелевскую премию по химии за «Природу химической связи».
После атомной бомбардировки Хиросимы и Нагасаки Полинг стал антивоенным активистом и даже имел из-за этого небольшие неприятности со стороны Госдепартамента. А в 1958 г. он добился прекращения ядерных испытаний, передав Генеральному Секретарю ООН петицию, подписанную более чем 11 000 ученых. В 1963 г. Полинг получил Нобелевскую премию мира — почему-то за 1962 г.
Вычислительные проблемы
Но может быть, причиной падения интереса к квантовой химии стал вовсе не фашизм и не война? Может быть, дело было в самой квантовой химии?
Квантовые химики знают, как быстро растет объем вычислений с размером молекулы даже при расчетах обычным методом Хартри—Фока. А метод-то сам по себе довольно неточный и едва годится даже для качественных оценок. Улучшить эти оценки можно, добавляя к рассчитанной волновой функции другие конфигурации, выбранные определенным способом. Это еще сильнее осложняет жизнь. Очень быстро стало понятно, что ничего интересного с разумной точностью вручную посчитать не удастся.
После войны, однако, квантовохимические расчеты возобновились — у ученых появились компьютеры. Развиваться стали как неэмпирические, так и полуэмпирические методы.
В 1951 г. метод Хартри—Фока был скомбинирован с представлением молекулярных орбиталей как линейных комбинаций атомных орбиталей. Получившиеся уравнения оказались весьма удобными для решения с помощью компьютеров. И все-таки просто переложить тяжелые и нудные вычисления на теплые ламповые плечи оказалось недостаточно. Пришлось обратиться к полуэмпирическим методам.
Метод Хюккеля был предложен в 1930 г., и ничего другого к началу 1950-х гг. в распоряжении квантовых химиков не было. В 1953 г. Рудольф Паризер, Роберт Парр и Джон Попл разработали новый полуэмпирический метод, впоследствии названный их именами. Как оказалось, метод довольно хорошо работает и имеет достаточно строгое обоснование. Однако, как и метод Хюккеля, метод ППП применим только к π-электронным системам — сопряженным углеводородам, красителям и т.п. Поэтому были разработаны более универсальные методы, включающие все валентные электроны.
Впрочем, универсальность полуэмпирических методов довольно-таки эфемерная. Параметризуют их под какое-то одно свойство: теплоту образования, например, или геометрию молекулы, или энергию ионизации. Пока расчет не выходит за рамки дозволенных свойств — все ОК. Но стоит попытаться посчитать что-то незапланированное — и попадешь пальцем в небо.
У неэмпирической квантовой химии проблем с универсальностью нет, но есть проблемы со стоимостью расчетов. Казалось бы, возьми компьютер помощнее — считай да радуйся. Но в квантовой химии отчетливее всего видно, что важен не столько мощный компьютер, сколько эффективно написанный код, а в последние годы — не просто эффективно написанный, но и хорошо распараллеленный. И все потому, что размер задачи растет быстрее, чем быстродействие компьютеров. Главное направление современной квантовой химии — разработка эффективных параллельных алгоритмов, реализующих наиболее продвинутые и точные методы.
Поначалу каждый теоретик писал программы для себя, но в 1970 г. Джон Попл с сотрудниками сделали Gaussian — первую квантовохимическую программу не для программистов, а для исследователей. Простота и удобство ее интерфейса до сих пор остаются образцом для квантовохимического софта. Программа поддерживается до сих пор и регулярно обновляется, чтобы включить новые, недавно появившиеся методы. Неудивительно, что практически в любой лаборатории, где занимаются квантовохимическими расчетами, есть (легальная или не очень) копия «Гауссиана» . Все квантовохимические программы, появившиеся после, вольно или невольно сравнивают с «Гауссианом»:
- по удобству интерфейса (конкурентов нет);
- по быстродействию и распараллеливанию (а тут как раз достойных конкурентов много);
- по реализации наиболее продвинутых или наиболее востребованных методов (тоже достойных конкурентов хватает).
История Gaussian’а не обошлась без скандала. С 1987 г. правами на программу владеет Gaussian Inc. (до этого программа была некоммерческой и свободно распространялась через Quantum Chemistry Program Exchange). В какой-то момент условия выдачи лицензии за версию программы стали включать пункт, запрещающий пользоваться «Гауссианом» тем, кто участвует в разработке конкурирующего софта, — подозреваемым конкурентам просто отказывали в продаже лицензии. Запрет толковался владельцами прав достаточно широко — от индивидуальных исследователей и небольших групп до целых факультетов и университетов. Тем временем сам Джон Попл в 1991 г. то ли ушел из Gaussian Inc., то ли его «ушли», но конфликт хоть и пожилого, но весьма активного профессора с бывшим учеником, а ныне владельцем Gaussian Inc. Майком Фришем имел место. Попл перешел в Q-Chem Inc., и принимал участие в разработке программы Q-Chem.
В 1998 г. Джон Попл вместе с Вальтером Коном (помним, он в 1964 г. разработал теорию функционала плотности) получил Нобелевскую премию по химии за развитие вычислительных методов.
Когда молекул много
Химическая связь — это, конечно, замечательно. Но еще в 1873 г. Ян Дидерик Ван-дер-Ваальс предположил, что между молекулами тоже существует взаимодействие. Оно достаточно слабое, так что не приводит к образованию химических связей [14], но достаточно сильное, чтобы вызвать отклонение поведения газов от идеального, а также чтобы способствовать конденсации газа в жидкость и кристаллизации жидкости. А когда полвека спустя стали изучать структуру и свойства биомолекул, то обнаружили, что практически все процессы, идущие с их участием, основаны на нековалентных межмолекулярных взаимодействиях. Ван-дер-Ваальс предположил, что эти взаимодействия имеют электростатическую природу.
Для электростатических взаимодействий (двух точечных зарядов, точечного заряда и диполя, двух диполей и т.п. — см. рисунок 12) формулы были известны уже в XIX в. Однако к взаимодействию молекул их применил в 1915 г. Виллем Хендрик Кеесом.
 а
а б
б в
в г
г д
д е
еРисунок 12. Электростатические взаимодействия. Взаимодействия: а — ион—ион; б — ион—постоянный диполь; в — взаимодействие постоянных диполей; г — ион—наведенный диполь; д — постоянный диполь—наведенный диполь; е — взаимодействие флуктуирующих диполей.
сайт web.mst.edu
Постоянные диполи могут поляризовать молекулы — так образуются наведенные (индуцированные) диполи. Индукционное взаимодействие изучал Петер Дебай в 1920–1921 гг.
Изучение взаимодействия наведенных диполей между собой и с мгновенными (флуктуирующими) диполями оказалось более сложной задачей, но ее решил в 1930 г. уже известный нам Фриц Лондон.
Водородная связь — это притяжение между атомом водорода или группой H—X (где X — более электроотрицательный атом, чем H) и другим электроотрицательным атомом или группой в той же или другой молекуле (рис. 13). Понятие водородной связи ввели в 1912 г. Том Мур и Томас Винмилл для аминов в водных растворах. Водородную связь в уже привычном нам виде для воды описали Венделл Латимер и Ворт Родебуш в 1920 г. В 1937 г. Карл Вольф ввел понятие супермолекулы для описания водородно-связанного димера уксусной кислоты, таким образом положив начало супрамолекулярной химии. Водородные связи стабилизируют α-спирали и β-листы в белках, удерживают пары оснований в нуклеиновых кислотах.

Рисунок 13. Водородная и ионная связи. а — Водородная связь — особый случай взаимодействия собственных диполей в молекуле. б — Водородные связи между парой азотистых оснований в ДНК. в — Ионная связь — особый случай взаимодействия заряженных групп в молекуле.

Рисунок 14. Пример солевых мостиков в тетрамере гемоглобина.
сайт wikimedia.org
Частным случаем взаимодействия зарядов является ионная связь (рис. 13в). Чаще всего ионную связь упоминают в связи с ионными кристаллами, типа поваренной соли NaCl, но это не единственное ее проявление. В частности, благодаря ионной связи образуются солевые мостики в белках, стабилизирующие их структуру и обеспечивающие функциональность (рис. 14).
Электростатические, поляризационные и дисперсионные силы — это силы притяжения между молекулами. Они проявляются на больших расстояниях. А на малых расстояниях (т.е. при сильном сближении) молекулы отталкиваются. А причина этому — сугубо квантовое явление, называемое обменным, или Паулиевским, отталкиванием. Принцип Паули гласит, что в одной точке пространства не могут находиться два электрона с одинаковыми характеристиками, поэтому полностью заполненные электронные оболочки будут отталкиваться. На определенном межмолекулярном расстоянии притяжение и отталкивание уравновешивают друг друга.
Людям, далеким от квантовой физики, Вольфганг Паули известен не своими научными заслугами, а «эффектом Паули». Известно, что присутствие теоретика в экспериментальной лаборатории негативно влияет на работу приборов. Явление было открыто на примере самого Паули и неоднократно подтверждено. Чем круче теоретик, тем больше масштаб производимых разрушений.
Снова шарики и палочки
Итак, получается, что чем больше размер задачи, тем больше объем вычислений. Выходит, что с мечтой Полинга о теоретическом предсказании структуры белков можно распрощаться? Так, да не так.
Действительно, предсказывать структуру белков или расшифровывать рентгеновские дифрактограммы из первых принципов невозможно до сих пор. Однако Полинг и другие исследователи успешно делали это — строили модели белков и нуклеиновых кислот. Как?
Помните шаростержневые модели, которыми пользовался еще Гофманн в XIX веке? С появлением в 1923 г. рентгеновской кристаллографии органических соединений стало ясно, что структурные формулы — не абстрактная картинка, а вполне себе реалистичная модель, действительно отражающая строение молекулы. Правда, формула нарисована на плоскости, а атом углерода, как выяснилось, действительно имеет тетраэдрическую координацию. Более того, выяснилось что связи, изображаемые одной, двумя или тремя палочками (одинарные, двойные, тройные) имеют свою характерную длину: 1,5 Å, 1,3 Å и 1,1 Å. Шаростержневые модели пришлось уточнить с учетом гибридизации валентных орбиталей атома (спасибо, Полинг): sp3-гибридные атомы стали изображать шариками с палками-валентностями, торчащими по углам тетраэдра, в sp2-гибридных три палки торчали по углам равностороннего треугольника, в sp-гибридных атомах две палки-валентности торчали в противоположные стороны. Палки, изображающие кратные связи, делали пропорционально короче, чем палки—одинарные связи. Вот в такое «лего» и играли структурщики.
Рентгеновская кристаллография биомолекул началась с работ Дороти Кроуфут Ходжкин, которая расшифровала структуры холестерина (1937 г.), пенициллина (1946 г.), витамина B12 (1956 г.) и инсулина (1969 г.). За эти работы в 1964 г. она получила Нобелевскую премию.
Белки — молекулы гораздо более крупные, менее регулярные и очень плохо кристаллизующиеся. Однако и их структуру удалось расшифровать Джону Коудери Кендрю и Максу Фердинанду Перуцу (Нобелевская премия 1962 г.). Позже для структурных исследований белков стали применять ядерный магнитный резонанс (Ричард Эрнст, Нобелевская премия 1991 г. по химии, и Курт Вютрих, Нобелевская премия 2002 г. по химии). Сейчас двумерная ЯМР спектроскопия конкурирует с кристаллографией по количеству расшифрованных структур и имеет все шансы обогнать последнюю, поскольку может работать с белками, которые не кристаллизуются.
Нуклеиновые кислоты, в частности, ДНК кристаллизуются гораздо лучше белков. Но расшифровка структуры ДНК превратилась в целый детектив со шпионажем и погонями, как это описал в своей книге «Двойная спираль» [15] Джеймс Уотсон (Нобелевская премия 1962 г. по физиологии и медицине вместе с Фрэнсисом Криком и Морисом Уилкинсом) .
Я намерено не пересказываю историю открытия двойной спирали, отправляя читателя к книге Уотсона.
Впечатляющие расшифровки структуры белков и нуклеиновых кислот, выполненные в 1950–1960 гг., были сделаны с помощью тех самых шаростержневых моделей (рис. 15).

Рисунок 15. Шаростержневая модель ДНК, построенная Уотсоном и Криком. Жесткие азотистые основания вырезаны из листа металла, гибкий сахарофосфатный остов построен из палочек на шарнирах.
сайт wikimedia.org
В 1946 г. Террел Хилл и Франк Вестхеймер независимо предложили выражение для межатомного потенциала в молекуле, включающего гармонические вклады для связей и валентных углов (модель «шариков на пружинках») и потенциал Леннард-Джонса для невалентных взаимодействий. Эта модель годилась для жестких молекул. С добавлением торсионного вклада (вращения вокруг одинарных связей) модель стала применимой к нежестким молекулам, а с добавкой электростатического вклада — и к молекулам, содержащим заряженные группы, в частности, к белкам (рис. 16). Все параметры этих потенциалов (такие как равновесное положение и жесткость «пружинок») подбирали так, чтобы воспроизводить экспериментальные рентгеновские структуры. Такие потенциалы называли эмпирическими (в отличие от полуэмпирической квантовой химии, где часть параметров подбирали под эксперимент, а часть — вычисляли). Оптимальную структуру молекулы находили, минимизируя стерическую энергию.

Рисунок 16. Компоненты молекулярного силового поля.
сайт wikimedia.org
С увеличением доступности компьютеров расчеты структуры молекул с помощью силовых полей (т.н. молекулярная механика) стали достаточно популярными в значительной степени благодаря работам Нормана Аллингера, автора модели поля MM2 и соответствующей программы.
Следующим шагом стал переход от поисков оптимальной структуры (минимума стерической энергии) к динамике молекул. В каждый момент времени рассчитывали силы, действующие на атомы, и вычисляли траектории частиц. Молекулярная динамика позволила получать термодинамические параметры системы или наблюдать на модели протекание физических и химических процессов [16].
Первые работы по молекулярной динамике были сделаны в 1955 г. Э. Ферми, Дж. Паста, С. Уламом, в 1959 г. Б. Алдером и Т. Уэйнрайтом, в 1960 г. Дж. Гибсоном с сотрудниками и в 1964 г. А. Раманом. Конечно, это были еще не биомолекулы, а просто динамика жестких сфер (атомов) в ван-дер-ваальсовом потенциале — атомных жидкостей и кристаллов. Все расчеты проводили на компьютере.
Одним из первых, кто применил молекулярную динамику для моделирования ДНК и белков , был Майкл Левитт в начале 1970-х гг. (Нобелевская премия по химии 2013 г. вместе с Арье Варшелом и Мартином Карплусом) [17], [18].
Для моделирования динамики биомолекул, особенно если требуется «отследить» в реальном времени довольно длительный процесс типа фолдинга крупных белков, необходимы огромные вычислительные мощности. До недавнего времени выйти за микросекундный масштаб не хватало сил даже у суперкомпьютеров, но правильное вложение частного капитала позволило преодолеть миллисекундный рубеж (необходимый минимум для фиксации белковых сворачиваний-разворачиваний) и построить модели работы ряда белков. О достижениях миллиардера Дэвида Шоу и его суперкомпьютера Anton можно прочитать здесь: «Миллисекундный барьер взят!» [19] и «Калиевый канал in silico» [20]. О прелестях и перспективах «сухой» биологии в целом рассказывает статья «Я б в биоинформатики пошёл, пусть меня научат!» [21], а о таком ее частном и жутко интересном направлении, как драг-дизайн, — материалы «Драг-дизайн: как в современном мире создаются новые лекарства» [22] и «Виртуальные тропы реальных лекарств» [23]. — Ред.
Как скрестить ежа с ужом
Молекулярная механика в силовых полях — штука хорошая и позволяет решить множество задач, касающихся структуры биомолекул и отчасти их функций. Но далеко не все процессы поддаются решению с помощью молекулярной механики. Так, химические реакции, приводящие к образованию или разрыву ковалентных связей, не моделируются традиционными силовыми полями, где связи между атомами считаются фиксированными. Процессы, протекающие с участием возбужденных состояний, также не подвластны традиционным силовым полям.
Химические реакции и фотопроцессы моделируют с помощью квантовой механики — неэмпирической или полуэмпирической [24]. Однако, как мы помним, биомолекулы слишком велики и не влезают в квантовую химию даже на самых современных суперкомпьютерах. Выход нашли Арье Варшел, Майкл Левит и Вальтер Тиль в 1976 г.
Обычно исследователей интересует реакционный центр — это ограниченная область большой молекулы, где происходит все интересное: химическая реакция или поглощение/испускание света. Этот реакционный центр моделируют методами квантовой химии. Все остальное — это окружение, и его моделируют методами молекулярной механики, поскольку его роль в исследуемом процессе вторична. Такая схема называется QM/MM (Quantum Mechanics/Molecular Mechanics) (рис. 17). В вычислительном отношении такая задача вполне подъемна, возможности расчета лимитируются только размером QM-части. Основную проблему — как аккуратно сшить QM- и MM-части — удалось решить авторам метода.

Рисунок 17. Принцип разделения системы на квантовомеханическую и молекулярно-механическую области.
QM/MM успешно комбинируется с молекулярной динамикой, чтобы получить реалистичные картины химических реакций и фотопроцессов в активном центре.
Закончилось ли на QM/MM развитие теоретических методов для моделирования биомолекул? Определенно нет. С увеличением мощности суперкомпьютеров стало возможно использовать в QM-части сложные продвинутые методы квантовой химии. Работают также над улучшенным описанием взаимодействия QM- и MM-частей.
А что, если в системе нет какого-то одного важного активного центра, зато есть много однотипных участков, ведущих себя схожим образом? Например, аминокислоты при упаковке белка, азотистые основания в ДНК или сахара в углеводах? Для этого есть методы т.н. огрубления (coarse-graining) [25]. Уже знакомые нам А. Варшел и М. Левитт в тех же работах середины 1970-х предложили схему, согласно которой целые аминокислотные остатки заменяются некими жесткими фигурами (помните вырезанные из железных листов азотистые основания в модели ДНК Уотсона и Крика?). Для жестких фигур задают потенциалы взаимодействия с прочими компонентами системы и в динамике получают траектории (рис. 18). Такие огрубленные расчеты позволяют гонять динамику на больших временах или исследовать системы большего размера. При необходимости огрубленный фрагмент можно «оживить» обратно, вернув ему атомную структуру, и посмотреть на него более подробно.

Рисунок 18. Пример огрубления в модели нуклеиновой кислоты.
сайт www.phas.ubc.ca
Методы моделирования, разработанные для биомолекул, давно уже перекочевали и в обычную химию, и в науку о материалах, где успешно помогают решать задачи, связанные с окружением активного центра или морфологией материала.
Вся эта поразительная история от первой реалистичной модели атома до моделирования структуры и функций огромных биомолекул и белковых комплексов уложилась меньше чем в столетие!
Литература
- van’t Hoff J.H. (1874). A suggestion looking to the extension into space of the structural formulas at present used in chemistry and a note upon the relation between the optical activity and the chemical constitution of organic compounds. Archives neerlandaises des sciences exactes et naturelles. 9, 445–454;
- Perrin J.-B. (1926). Discontinuous structure of matter. Сайт Nobelprize.org;
- Lewis G.N. (1916). The atom and the molecule. J. Am. Chem. Soc. 38, 762–785;
- Глинка Н.Л. Общая химия: Учеб. пособие для вузов / Под ред. Ермакова А.И. М.: ИНТЕГРАЛ-ПРЕСС, 2003. — 728 с.;
- Атомно-силовая микроскопия: увидеть, прикоснувшись;
- Pauling L. (1931). The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Amer. Chem. Soc. 53, 1367–1400;
- Pauling L. (1931). The nature of the chemical bond. II. The one-electron bond and the three-electron bond. J. Amer. Chem. Soc. 53, 3225–3237;
- Pauling L. (1932). The nature of the chemical bond. III. The transition from one extreme bond type to another. J. Amer. Chem. Soc. 54, 988–1003;
- Pauling L. (1932). The nature of the chemical bond. IV. The energy of single bonds and the relative electronegativity of atoms. J. Amer. Chem. Soc. 54, 3570–3582;
- Pauling L. and Wheland G.W. (1933). The nature of the chemical bond. V. The quantum-mechanical calculation of the resonance energy of benzene and naphthalene and the hydrocarbon free radicals. J. Chem. Phys. 1, 362–374;
- Pauling L. (1939). The nature of the chemical bond and the structure of molecules and crystals. Cornell University Press, 1960. — 664 p.;
- Проблема фолдинга белка;
- На заре молекулярной графики;
- Роль слабых взаимодействий в биополимерах;
- Уотсон Д. Двойная спираль. Воспоминания об открытии структуры ДНК. М.: Мир, 1969. — 152 с.;
- Молекулярная динамика биомолекул. Часть I. История полувековой давности;
- Жизнь — это компьютер;
- «Виртуальная» Нобелевская премия по химии (2013);
- Миллисекундный барьер взят!;
- Калиевый канал in silico;
- Я б в биоинформатики пошёл, пусть меня научат!;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Виртуальные тропы реальных лекарств;
- Порог чувствительности зрительного восприятия;
- Kmiecik S., Gront D., Kolinski M., Wieteska L., Dawid A.E., Kolinski A. (2016). Coarse-grained protein models and their applications. Chem. Rev. 116, 7898–7936.



Комментарии
0Чтобы оставить комментарий, необходимо
войти