
Связь клеток — связь времен
28 января 2021
Связь клеток — связь времен
- 3070
- 0
- 10
-
Автор
-
Редакторы
Статья на конкурс «Био/Мол/Текст»: Всем известно, что наш организм состоит из клеток. Триллионов клеток! Но задумывались ли вы, что скрепляет все эти клетки вместе и делает нас такими, какие мы есть? В единый организм массу клеток объединяют компоненты соединительной ткани, а конкретней, внеклеточного матрикса. Поговорим о том, какую роль внеклеточный матрикс играет в организме, а также о том, как человечество может использовать свои знания о нем для достижения столь желанной для многих людей цели — долгожительства.

Конкурс «Био/Мол/Текст»-2020/2021
Эта работа опубликована в номинации «Свободная тема» конкурса «Био/Мол/Текст»-2020/2021.
Генеральный партнер конкурса — ежегодная биотехнологическая конференция BiotechClub, организованная международной инновационной биотехнологической компанией BIOCAD.

Спонсор конкурса — компания SkyGen: передовой дистрибьютор продукции для life science на российском рынке.
Спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
«Книжный» спонсор конкурса — «Альпина нон-фикшн»
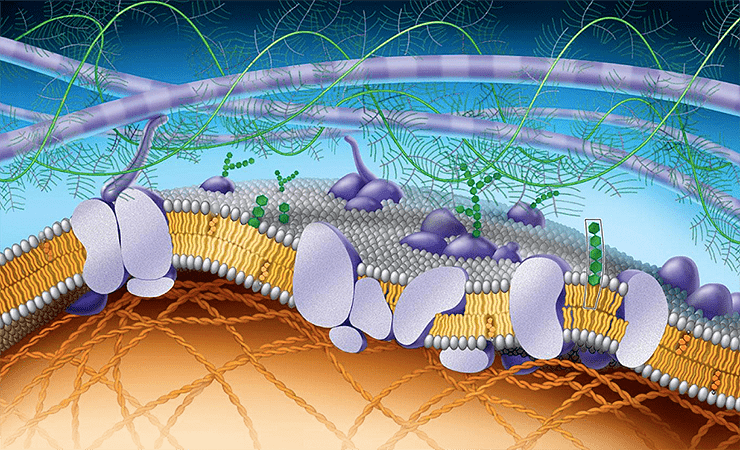
Что такое внеклеточный матрикс и зачем он нужен?
Внеклеточный матрикс находится в основе соединительной ткани, которая, помимо всем известной функции каркаса нашего организма, выполняет и еще одну важную функцию — коммуникации или общения клеток друг с другом.
Давайте посмотрим на устройство жировой ткани в качестве примера. Главный ее компонент — жировые клетки, адипоциты. Их подавляющее большинство, но, как мы уже знаем, еще есть межклеточный матрикс, а также немного клеток-фибробластов (отвечают за обновление матрикса), предшественники адипоцитов (мало ли, понадобится новая клетка), иммунные клетки (в основном речь о макрофагах, которые любят пожирать ненужное) и какое-то количество нервных окончаний, артерий и вен. Схожее строение имеют многие другие ткани в человеческом организме.
Из чего же состоит матрикс? Его основу формируют гиалуроновая кислота, гликопротеины и протеогликаны. Гиалуроновая кислота — полимер, который состоит из остатков D-глюкуроновой кислоты и D-N-ацетилглюкозамина. Помимо межклеточного матрикса она является основным компонентом слюны и синовиальной жидкости и содержится в биологических смазках. Гликопротеины и протеогликаны имеют сложное строение: они состоят из белка и олигосахаридной цепи, однако по массе у протеогликанов углеводов существенно больше, чем у гликопротеинов. К данной группе белков относятся всем известный коллаген, который составляет основное большинство молекул матрикса, эластин, фибрин, компоненты базальных мембран (ламины) и еще некоторые другие молекулы.
Компоненты матрикса постоянно обновляются: старый матрикс расщепляется группой специальных ферментов (матриксными металлопротеиназами), а новые компоненты синтезируются специализированными клетками (например, фибробластами и хондроцитами). Некоторые компоненты матрикса обновляются быстро, но некоторые, например, коллаген и эластин, являются долгоживущими. Период полураспада коллагена в межпозвоночных дисках составляет целых 95 лет [1]. Поэтому логично, что они претерпевают различные химические изменения и накапливают повреждения.
Остановимся немного на металлопротеиназах. Они разрушают белки матрикса, тем самым позволяя ему обновляться и перестраиваться. В каталитический центр этих ферментов входит металл, чаще всего это цинк, реже — кобальт. Как и другие ферменты, они подразделяются на две группы — могут быть экзопептидазами (отщепляют аминокислоты от конца белка) и эндопептидазами (расщепляют белок внутри пептидной цепи). Они участвуют, например, в важных процессах жизнедеятельности мышц и ангиогенезе.
Обновление матрикса может помогать в заживлении ран и ожогов, в связи с чем ученые активно исследуют клетки, способные производить коллаген и эластин — основные белки матрикса. Главную роль здесь играют дермальные фибробласты. Экспериментально было показано, что фактор роста фибробластов 1 стимулирует развитие собственной ткани организма и помогает нанесенной ране эффективно закрыться (тем самым блокируя инфекцию и уменьшая образование рубцов). Использование фактора роста фибробластов, стимулирующего активность этих клеток, — более эффективно для заживления ткани, чем применение существующих тканевых герметиков, так как в результате образуется очень прочный коллаген типа I [2]. Он состоит из нескольких нитей, связанных в прочные фибриллы, и имеет уникальную трехспиральную структуру, которая повышает его целостность (рис. 1). Весьма перспективными являются исследования в области стимулирования работы фибробластов для выработки коллагена, ведь это может помочь не только в заживлении нанесенных ран, но и при реставрации стареющих тканей за счет синтеза новых компонентов межклеточного матрикса.

Рисунок 1. Структура коллагена I типа. Красная линия — коллаген I-альфа-I; синяя линия — коллаген I-альфа-II.
Поскольку матрикс в организме играет не только структурную роль, важно сказать, за счет чего осуществляется другая его важная функция — коммуникативная.
Основное место здесь занимают селектины — гликопротеины, которые пронизывают мембрану клетки и участвуют в процессе связывания клетки с матриксом и другими клетками. По своей природе они принадлежат семейству белков лектинов (отчего и сходство в названии), а значит, могут «склеивать» остатки углеводов на клеточной поверхности. Важно помнить, что селектины, как и другие лектины, участвуют не только в связывании клеток, но и во внутриклеточном сигналинге.
Все селектины активируются, прикрепляясь к особым гликопротеиновым и гликолипидным лигандам на поверхности клетки. Также важно знать, что они играют важную и хорошо изученную роль в рекрутинге («вербовке») и миграции лейкоцитов в места воспаления. Это делает селектины незаменимыми не только для связывания клеток между собой, но и для формирования иммунного ответа. Почему это для нас важно, расскажем немного позднее.
Нарушения в процессе активации селектинов (например, повышенный уровень их с лигандами), могут привести к обострению различных заболеваний, включая атеросклероз, рестеноз, тромбоз глубоких вен и запуск метастазирования опухолей. А значит, в некоторых случаях искусственная блокировка селектинов может облегчить течение заболевания [4].
Как связаны старение организма и изменения во внеклеточном матриксе?
Возрастное ремоделирование матрикса
Свойства матрикса достаточно сильно влияют на работу клеток, в частности, на их развитие и дифференцировку. Клетки «чувствуют» степень жесткости матрикса при помощи различных механорецепторов. Поэтому растущая с возрастом жесткость матрикса и изменения в его структуре влияют на работу клеток, на их способность к адгезии, на миграцию стволовых клеток и на другие аспекты их поведения [5].
Негативные изменения в матриксе связывают в первую очередь с таким химическим процессом, как гликирование белков (рис. 2), и в меньшей степени с накоплением рацематов аминокислот [6]. Эти процессы довольно опасны. Накопление рацематов межклеточного матрикса в нервной ткани мозга может, например, приводить к нарушению его функционирования [5], что, вероятно, и происходит при когнитивных заболеваниях пожилых людей.
- Рацемат
- смесь двух оптических вариантов молекул, которые представляют собой зеркальные отражения друг друга, несовместимые в пространстве.
Гликирование белков может значительно изменять свойства коллагена I типа — он становится устойчив к ферментативному ремоделированию и из-за этого между волокнами коллагена формируются сшивки, что увеличивает жесткость матрикса [1]. Такое увеличение связывают с развитием ряда возрастзависимых заболеваний: повышением толщины и жесткости стенок сосудов [8], возрастными изменениями сердца [9], онкологическими [10] и нейродегенеративными заболеваниями [11].

Рисунок 2. Схематичное изображение процесса гликирования белков
Сам процесс гликирования представляет собой неферментативное присоединение моносахаридов (глюкозы, фруктозы или их производных) к свободной аминогруппе белка. Химическое названия данной реакции — реакция Майара.
Биологическое предназначение гликирования — индукция синтеза провоспалительных цитокинов [12], регуляция апоптоза [13], участие в регуляции времени жизни белков [14].
Однако в целом это процесс для организма не очень полезный (зато весьма полезен при жарке стейка!), так как, например, в результате него образуется большинство повреждений тканей при сахарном диабете [15]. Кроме этого гликирование может приводить к нарушению функций и структуры митохондрий и окислительному стрессу [16]. Повышенное содержание продуктов гликирования было определено в нервных клетках пациентов, страдающих болезнью Альцгеймера [17], что указывает нам на участие рассматриваемого процесса в патогенезе этой тяжелой болезни.
Очень важно в контексте гликирования белков межклеточного матрикса упомянуть о таком явлении, как инфламмэйджинг.
Инфламмэйджинг — это развитие хронического воспаления низкого уровня в пожилом возрасте. Оно опасно тем, что усугубляет течение таких сенильных заболеваний, как атеросклероз, диабет второго типа, болезнь Альцгеймера и других патологий [19]. Гликированные продукты белков межклеточного матрикса активно привлекают макрофагов — значимых участников воспалительных процессов. В результате концентрация макрофагов в ткани увеличивается, и в ней развивается повышенный уровень воспаления [20].
Так как же быть?
Поскольку проблема повреждения тканей из-за протекающего в них процесса гликирования проявляется в контексте многих заболеваний, ученые активно ищут пути ее решения. Уже идут подбор и разработка веществ, которые могли бы ингибировать этот процесс или поворачивать его вспять. Рассмотрим некоторые из них.
ДНК-аптамеры представляют собой олигонуклеотиды, нацеленные на блокаду рецепторов , связывающихся с конечными продуктами гликирования (КПГ) белков матрикса. Их предполагается использовать для восстановления поврежденных в результате этого процесса органов [21].
Рецепторы RAGE представляют собой иммунные рецепторы из семейства иммуноглобулинов и активируются в ответ на КПГ, амилоиды и продукты клеточной гибели. RAGE-рецепторы обильно производятся в жировой ткани и участвуют в регуляции инсулинового сигналинга. Существует гипотеза, что первоначальная роль RAGE-рецепторов в эволюции гоминидов была связана с формированием экономного фенотипа с ускоренным набором жировой массы, что противодействовало голодной смерти.
Способность блокировать RAGE-рецепторы обнаружена и у некоторых искусственно созданных аминокислот [22], а также у дейтерированных полиненасыщенных жирных кислот [23], хебуловой кислоты [24], кверцетина [25] и молекулы GLY-230 [26]. Однако у такого подхода есть недостатки — в низких концентрациях эти блокаторы оказываются неэффективными, а в высоких — токсичными. Систему, однако, можно модифицировать, подбирая комбинации компонентов, которые в совокупности будут действовать также эффективно, но из-за более низких концентраций будут менее ядовиты.
Одна группа ученых предложила генетически модифицировать фибробласты, чтобы они секретировали вещество амадориазу . Это могло бы предотвратить появление таких продуктов гликирования, как, например, глюкозепан. Глюкозепан — основной связующий компонент [27], образующийся при гликировании белков. Его в 45 раз больше, чем, например, реактивных дикарбониловых конечных продуктов гликирования. Его высокое содержание как раз может быть причиной того, что искусственно созданные вещества для блокирования рецепторов конечных продуктов гликирования не работают, ведь они в основном направлены на предотвращение образования не глюкозепановых сшивок. Совсем недавно описанные антитела к глюкозепану должны значительно облегчить ученым исследования этого распространенного связующего компонента при гликировании [28].
Амадориаза — фермент, действующий на низкомолекулярные соединения аминокислот с сахарами. Был обнаружен у грибов и бактерий [29].
Компания «Альтеон» даже создала первое лекарство против различных сшивок в межклеточном матриксе — «Алагебриум». Оно способно разрезать сшивки, образованные при помощи α-дикетона. Однако лекарство не имело большого успеха, поскольку таких сшивок в матриксе оказалось не слишком много, и основная проблема повышения его жесткости с помощью препарата не решалась.
Совсем недавно появилась статья, в которой предлагается использовать 4-фенилбутират натрия в качестве агента против гликирования [30]. Механизм его действия точно неизвестен, однако предполагается, что он может связываться с альбумином и предотвращать его взаимодействие с глюкозой, что и является начальным этапом гликирования. Так 4-фенилбутират натрия становится потенциальным участником на поле борьбы с нейродегенеративными заболеваниями, атеросклерозом, диабетом, гиперлипидемией и другими возрастзависимыми недугами.
Очень интересный способ «отменить» гликирование показала группа исследователей, которая предложила использовать фермент под названием MnmC для обратного гликированию процесса. Этот фермент в природе участвует в процессе модификации тРНК бактерий [31]. Возможно, именно такого рода открытие и станет отправной точкой в терапии болезней, в патогенезе которых играет роль процесс гликирования.
Круговая порука или ренин-ангиотензин-альдостероновая система
Однако выяснилось, что с процессами в изменении структуры матрикса также связана активность РААС — ренин-ангиотензин-альдостероновой системы (рис. 4).
РААС отвечает за регуляцию давления и распределения крови в организме человека. Краткий механизм ее действия таков: гормон ренин (вырабатывается в почках) отщепляет от гормона ангиотензиногена (вырабатывается в печени) кусочек, в результате чего образуется неактивный ангиотензин I. В клетках эндотелия легочных капилляров ангиотензин I гидролизуется ангиотензинпревращающим ферментом, который отщепляет от него еще кусочек. В результате образуется активный ангиотензин II. Он — мощный вазоконстриктор, то есть способен влиять на сокращение сосудов. Таким образом образуется цепь реакций, которая регулируется большим количеством звеньев.

Рисунок 4. Схема работы РААС
Постоянная и чрезмерная активация РААС, например, при избыточном синтезе ангиотензиногена в печени, играет существенную роль в патологической гипертрофии и ремоделировании сердца при старении. Активация рецептора к антигиотензину II AT1, который представлен на поверхности клеток многих тканей (сердце, почки, нервная система и др.), вызывает чрезмерные ответы в кардиомиоцитах и синтез белка внеклеточного матрикса в фибробластах сердца. Последствия этих процессов печальны — функции сердца сильно страдают [32–36]. Кроме непосредственно изменения в структуре сердечной ткани, благодаря действию РААС могут индуцироваться различные клеточные ответы в сосудистой стенке, что приводит к прогрессированию атеросклеротического процесса — одной из серьезных проблем пожилых людей [37], [38].
Решение этой проблемы уже проглядывается — опыты с животными показали, что фармакологическое ингибирование компонентов РААС приостанавливает увеличение количества компонентов матрикса, приводящее к фиброзу, а значит, и патологические процессы в тканях сердца [39].
В фибробластах сердца ангиотензин II повышает синтез коллагена, фибронектина, ламинина и остеопонтина. Гладкомышечные клетки сосудов, стимулированные ангиотензином II, демонстрируют увеличение количества мРНК коллагена, фибронектина, ламинина и тенасцина. Помимо всего этого гормон также стимулирует синтез коллагенов, фибронектина и ламинина мезангиальными клетками в почках [39], [40].
Способность ангиотензина II стимулировать продукцию TGF-β , одного из главных профиброзных факторов, связывают с развитием возрастной сосудистой гипертрофии.
TGF-β — многофункциональный цитокин, который участвует в процессах клеточной дифференцировки и пролиферации, иммуном ответе, а также играет важную роль в регуляции сборки и ремоделирования внеклеточного матрикса.
TGF-β, продукцию которого стимулирует ангиотензин II, стимулирует синтез коллагенов, фибронектина и протеогликанов и выработку протеаз, которые ингибируют распад матрикса. Постоянная активация рецепторов TGF-β приводит к аномальному накоплению соединительной ткани, связанному с фиброзными патологиями почек и сосудов [42–44], Кроме того, продукция TGF-β может индуцироваться при увеличении количества рецепторов RAGE [45]. То есть повышение выработки этого цитокина может быть прямым следствием процессов гликирования. Возможно, если получится изобрести препарат, который «отменял» бы гликирование, то вред, причиняемый большими количествами TGF-β, тоже снизится [46–48].
Митохондрии и матрикс — продолжение истории
Работа митохондрий и матрикса тесно связаны друг с другом посредством работы цитоскелета.
Цитоскелет — внутриклеточный каркас, который регулирует форму клетки под влиянием внешних воздействий, поддерживает внутриклеточный порядок, участвует в движении клеток и в ряде других процессов.
Для обновления белков основных структур цитоскелета (микрофиламентов, промежуточных филаментов, микротрубочек), требуется энергия аденозинтрифосфата (АТФ). Поэтому митохондрии в клетке постоянно двигаются, собираясь в местах, где высока потребность в АТФ. В свою очередь, правильная организация скелета клетки очень важна для нормального функционирования митохондрий — они тесно взаимодействуют с ним для поддержания своей морфологии. Ангиотензин II нарушает нормальную организацию цитоскелетных филаментов, что негативно сказывается на работе митохондрий [49].
Как же взаимосвязаны изменение матрикса, нарушение работы цитоскелета и митохондрий? Поскольку ангиотензин II способен вызывать изменения в синтезе компонентов межклеточного матрикса, то изменяется и цитоскелет клетки.
Известно, что эндотелиальные клетки, находящиеся внутри более жесткого матрикса имеют более долго образующиеся, однако более прочные микротрубочки, в то время как клетки, находящиеся в менее жестком матриксе, образуют не такие прочные микротрубочки, которые растут быстрее [50]. Они же в свою очередь очень важны для функционирования митохондрий: при их «разборке» митохондрии теряют свою подвижность, что отрицательно сказывается на их структуре и нарушает энергетическую систему в клетке [51].
Еще одним из механизмов такого нарушения цитоскелета клетки, помимо антиогензина II, является регуляция реорганизации актиновых филаментов при помощи уже упомянутого выше цитокина TGF-β. Вызывая перегруппировки актиновых филаментов, он влияет на рост и дифференцировку клеток, так как в ядре запускается действие определенных транскрипционных факторов , [46–48].
Также TGF-β может вызывать дисфункцию митохондрий. Так, этот фактор инициирует остановку роста и приобретение фенотипов старения в эпителиальных клетках легких, снижая активность митохондриального комплекса IV — цитохром-С-оксидазы. Цитохром-C-оксидаза замыкает дыхательную цепь, перенося электроны на кислород. Снижение активности комплекса приводит к утечке электронов и образованию активных форм кислорода. Активные формы кислорода (АФК) — постоянно образующиеся в клетке метаболиты кислорода, а именно: его ионы, свободные радикалы и перекиси. Их особенность в том, что они очень высокореактивны за счет того, что на их внешнем электронном уровне присутствует неспаренный электрон. АФК в норме участвуют в иммунных реакциях и некоторых метаболических путях, но при избытке могут привести к окислительному стрессу клетки. Окислительный стресс потенциально является причиной некоторых старческих заболеваний, например, болезни Альцгеймера [56], атеросклероза [57], болезней повреждения митохондрий [58].

Рисунок 5. Транспорт TGF-β-рецепторов. Комплексы рецепторов TGF-β, находящиеся в областях мембран, которые образуют окаймлённые ямки (серая линия), интернализуются, возможно, под действием динеинов вдоль микротрубочек и локализуются в эндосомах. Фосфорилирование R-Smad стимулируется в эндосомах на основе действия эндоцитарного белка SARA, что приводит к транскрипционной сигнализации. Эндосомальные рецепторы могут рециркулировать обратно в плазматическую мембрану или переходить в лизосомы, где лиганд-рецепторный комплекс деградирует (красный крест). Комплексы рецепторов TGF-B, находящиеся в области мембраны, называются липидными рафтами (белая линия), связываются со Smad7 и Smurf убиквитинлигазами и интернализуются в кавеолы. Эти комплексы рецепторов переходят в лизосомы для последующей деградации, и участвуют ли они в передаче сигнала, остается неизвестным. Возможность взаимодействия между эндосомальным рецепторным пулом и кавеолярным рецепторным пулом не установлена и отмечена двойной стрелкой со знаком вопроса.
Совсем недавно ученые описали митохондриально-матриксный путь в развитии саркопении, одной из патологий, возникающих в пожилом возрасте. При саркопении происходят патологические изменения мышечных волокон, приводящие к атрофии скелетных мышц. Препаратов, эффективных в лечении саркопении, пока, к сожалению, не разработано. Известно, что в развитии саркопении важную роль играет нарушение функционирования митохондрий [59]. Такая дисфункция митохондрий приводит не только к энергетическому дефициту клеток мышц, но и может запускать апоптоз мышечных клеток и иннервирующих их двигательных нейронов [60]. Изменения внеклеточного матрикса, в частности, «старение» коллагена [61], [62] и увеличение активности матриксных металлопротеиназ [63], также, несомненно, участвуют в развитии саркопении, хотя точных молекулярных механизмов действия этих факторов на данный момент нет. Также пока что не было предложено и единого механизма, который связал бы эти возрастные изменения в одну модель,
Годом ранее команда А. Мелуан показала, что некий белок SPARC влияет одновременно и на изменения состава внеклеточного матрикса, и на функцию митохондрий в мышечных клетках [64].
Белок SPARC (аббревиатура переводится как «секретируемый белок, богатый цистеином») также известен как остеонетин и является кальций-связывающим матрично-клеточным гликопротеином [65].
В мышцах SPARC синтезируется при строительстве или заживлении мышечной ткани. А еще SPARC обладает способностью связываться с коллагенами разных типов, за счет чего он влияет на перестройку и формирование внеклеточного матрикса [66]. Что касается митохондрий, то этот белок влияет на их развитие путем взаимодействия с индуктором биогенеза митохондрий белком AMPK (протеинкиназы, активированной аденозинмонофосфатом) [67].
В другой свежей работе описана взаимосвязь нарушения функций митохондрий и вызванного этим переизбытка кальция с нарушением структуры внеклеточного матрикса в мышцах. Было бы удивительно, если бы наш «герой» ангиотензин II не поучаствовал вместе с дисфункцией матрикса и митохондрий в потере мышечной массы [68]. И это действительно так, что показано в работе японских исследователей [69]. Кроме того, не обошлось без участия белка SPARC. Схема их непростых взаимодействий представлена на рисунке 6.

Рисунок 6. Влияние белка SPARK на синтез внеклеточного матрикса и на репликацию и транскрипцию мтДНК. Условные обозначения: АМФ — аденозинмонофосфат; АТФ — аденозинтрифосфат; ВКМ — внеклеточный матрикс; AMPK — аденозинмонофосфаткиназа; ILK — интегрин-связанная киназа; GSK-3β — 3-бета-киназа гликогенсинтазы; PGC1α — 1-альфа-коактиватор рецептора гамма, активируемого пролифератором пероксисом.
Общая цепь событий может быть представлена таким образом: повышенная продукция ангиотензина II в организме → дисфункция митохондрий и окислительный стресс → нарушение гомеостаза кальция → нарушение структуры внеклеточного матрикса → дистрофия мышц → саркопения.
Когда стоит поберечь жир?
Жир порой бывает весьма полезен для организма. Его повреждение может приводить к очень неприятным последствиям. Давайте поговорим об этом более подробно.
Перекисное окисление липидов — это повреждение полиненасыщенных жирных кислот свободными радикалами, которое происходит в митохондриальных и клеточных мембранах. Двойные связи полиненасыщенных жирных кислот особенно чувствительны к воздействию свободных радикалов, так как легко «разрываются», присоединяя их, и образуют диальдегиды, пероксиды и другие продукты окисления. Все это запускает цепную реакцию окисления липидов. Перекисное окисление липидов происходит, в первую очередь, во внутренней мембране митохондрий, которая находится в активном контакте со свободными радикалами, а также в клеточной мембране нейронов. Этот процесс изменяет физические свойства мембран, их текучесть и работу электрон-транспортной цепи митохондрий. Продукты перекисного окисления токсичны и повреждают важнейшие долгоживущие молекулы — некоторые белки (гистоны, белки ядерных пор, структурные белки) и ДНК [71]. Ряд исследований указывает на количественную связь между процессом перекисного окисления липидов и образованием поперечных сшивок белков матрикса [72]. Так, например, один из продуктов перекисного окисления липидов, малондиальдегид, образует такое же количество сшивок с белками, как и глюкоза, что было показано в экспериментах in vitro [72]. Долгоживущие виды животных имеют в составе своих клеток меньше полиненасыщенных кислот, чем короткоживущие. В связи с этим можно предположить, что они не только в меньшей степени страдают от перекисного окисления липидов, но и имеют более замедленный, за счет уменьшения реакционной способности жирных кислот, процесс изменения белков внеклеточного матрикса. Из всего это следует, что процесс окисления жирных кислот отрицательно влияет на состояние внеклеточного матрикса, что, как мы уже знаем, приводит к неприятным последствиям — старению тканей, их фиброзу и нарушению их функций.
Тут на помощь нам могут прийти антиоксиданты. Основных механизмов их действия для предотвращения перекисного окисления липидов три:
- Предотвращение формирования активных перекисей.
- Ликвидация активных перекисей.
- Починка поврежденных в результате окисления липидов и вывод токсичных продуктов окисления из клетки.
Таким образом, в перспективе преодоления этой проблемы антиоксиданты могут оказаться действительно полезными в предотвращении некоторых признаков старения.
Почему ломит кости?
Наверняка вы слышали, что пожилые люди часто жалуются на то, что у них ломит кости. А вы задумывались, почему так происходит и как можно этого избежать? Удивительным образом тут тоже замешан внеклеточный матрикс.
Начнем издалека: в костях активно происходит кроветворение, или гемопоэз. В старении гемопоэтических стволовых клеток (ГСК) очень важную роль играет их микроокружение в костном мозге — так называемая ниша ГСК. Она состоит из мезенхимальных стволовых и эндотелиальных клеток. Так вот, старение мезенхимальных клеток этой ниши влияет и на ГСК. По мере прогрессирования этого процесса мезенхимальные клетки все слабее и слабее проявляют свои регенеративные способности. Некоторые исследователи показали, что это может даже привести к развитию воспаления и прогрессированию рака [73].
Но нам в данном случае важнее, что процессы старения стимулируют мезенхимальные стволовые клетки превращаться в жировые клетки (адипоциты) в ущерб развитию клеток костей [3], [7]. Это именно тот случай, когда жир совсем не полезен для здоровья организма. Активный адипогенез в старом костном мозге нарушает восстановление предшественников клеток крови, состав внеклеточного матрикса, образование кости и последующее восстановление переломов, что характерно для пожилых людей [41], а также способствует возникновению остеопороза [52], [53]. Как мы видим, нарушение функционирования внеклеточного матрикса внутри кости опосредованно способствует «ломкости» костей.
Подведем итоги
Внеклеточный матрикс — динамический внеклеточный компонент организма, который постоянно ремоделируется в ответ на различные стимулы и подвержен существенным изменениям в ходе старения организма. Его компоненты регулируют различные клеточные процессы, включая клеточную пролиферацию, выживание, дифференцировку и миграцию.
Внеклеточный матрикс состоит из множества белков. Самые распространенные из них — коллаген и эластин. Это довольно долгоживущие белки и, как следствие, особенно чувствительные к накоплению неферментативных посттрансляционных модификаций и фрагментации в результате ферментативного расщепления (как коллаген в старой коже). По современным представлениям, большинство продуктов посттрансляционных модификаций матричных белков образуют КПГ. Хотя остается еще много неясного о самом характере изменений, происходящих в матриксе... Например, продукт карбамилирования белков матрикса, гомоцитруллин, также часто встречается в стареющем матриксе и вносит свой вклад в изменение его функций. По мнению некоторых ученых, этот процесс может быть таким же весомым, как и гликирование [54]. Изменения матричных белков в процессе старения очень сильно влияют на функции матрикса, воздействуя на другие биологически активные молекулы. Так, активация сигнальных каскадов после взаимодействия КПГ с рецепторами RAGE приводит к многочисленным нарушениям функции клеток, в том числе к воспалительным и окислительным процессам с формированием замкнутых петель обратной связи. RAGE, как сейчас известно, участвует в развитии почти всех возрастных патологий, включая диабет, хроническое заболевание почек, сердечно-сосудистые заболевания, рак, болезнь Альцгеймера и Паркинсона.
Все эти изменения в старом внеклеточном матриксе могут напрямую влиять на его механическую и структурную роли. Как следствие, запускаются нежелательные процессы: истощение пула стволовых клеток, клеточное старение, нарушение межклеточной коммуникации, геномная нестабильность и дисфункция митохондрий. Кроме того, возрастная дисфункция матрикса напрямую связана с возрастными отклонениями и патологиями: нарушением целостности барьеров (кишечного и гематоэнцефалического), фиброзом, сердечно-сосудистыми и нейродегенеративными заболеваниями. Предполагается, что старение внеклеточного матрикса может быть даже более важным, чем старение самих клеток, так как внутри клетки, в отличие от матрикса, существуют более эффективные механизмы восстановления и удаления поврежденных белков и органелл.
Состояние внеклеточного матрикса можно считать биомаркером старения. Так, образование в нем сшивок и КПГ — признак того, что возраст наступает на пятки. Хотя на данный момент маркеры старения широко не используются, КПГ (глюкозепан и др.) довольно перспективны с точки зрения оценки возрастного состояния организма. Созданное недавно командой Дэвида Шпигеля антитело, связывающееся с глюкозепаном, также предоставляет еще один инструмент по идентификации в организме этого вида КПГ [55].
Какие пути решения предлагают ученые по противодействию негативным последствиям от старения матрикса? На сегодняшний день их несколько. Это, например, разработка комбинации ингибиторов КПГ, которые синергетически работают на разных стадиях их образования. Сюда могут входить хелаторы (препараты по связыванию избытка железа), соединения, содержащие О-ацетильную группу для защиты белков от гликирования и соединения с трансгликирующей активностью, амадорины и амадориазы.
Также необходимо сфокусироваться на разработке разрушителей основного КПГ во внеклеточном матриксе — глюкозепана (небольшую молекулу или фермент, способные проникать между фибриллами коллагена и достигать своей цели — сшивок КПГ). Кроме этого, нам нужны препараты, стимулирующие производство эластина, и «ловушки» провоспалительного рецептора RAGE (такие, как растворимые формы этого рецептора и др.). Ингибирование сигнальных путей, запускаемых жестким матриксом, по мнению ученых, также может быть полезным [70].
Достоверно ясно лишь одно: на сегодняшний день борьба со старением внеклеточного матрикса — это очень сложная и многофакторная задача, которая требует привлечения к себе большого внимания и существенного количества финансовых средств. Но не решив эту задачу, ученые-геронтологи и все мы вместе не сможем существенно продвинуться к более глобальной цели — победе над старением.
Литература
- Helen L. Birch. (2018). Extracellular Matrix and Ageing. Subcellular Biochemistry. 169-190;
- Sadanori Akita, Kozo Akino, Toshifumi Imaizumi, Akiyoshi Hirano. (2008). Basic fibroblast growth factor accelerates and improves second-degree burn wound healing. Wound Repair and Regeneration. 16, 635-641;
- MiJung Kim, ChanWha Kim, Yu Suk Choi, MinHwan Kim, ChanJeoung Park, Yousin Suh. (2012). Age-related alterations in mesenchymal stem cells related to shift in differentiation from osteogenic to adipogenic potential: Implication to age-associated bone diseases and defects. Mechanisms of Ageing and Development. 133, 215-225;
- Wei Luo, Hui Wang, Miina K. Öhman, Chiao Guo, Kate Shi, et. al.. (2012). P-selectin glycoprotein ligand-1 deficiency leads to cytokine resistance and protection against atherosclerosis in apolipoprotein E deficient mice. Atherosclerosis. 220, 110-117;
- Alexander Fedintsev, Alexey Moskalev. (2020). Stochastic non-enzymatic modification of long-lived macromolecules - A missing hallmark of aging. Ageing Research Reviews. 62, 101097;
- E. Man, M. Sandhouse, J Burg, G. Fisher. (1983). Accumulation of D-aspartic acid with age in the human brain. Science. 220, 1407-1408;
- J. Justesen, K. Stenderup, E.N. Ebbesen, Li Mosekilde, T. Steiniche, M. Kassem. (2001). . Biogerontology. 2, 165-171;
- Marie Paule Jacob. (2003). Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomedicine & Pharmacotherapy. 57, 195-202;
- Danisha M.D., Suman D., Connor J., Cerrone R.F., Krishna S. (2019). The Role of Osteopontin in Extracellular Matrix Remodeling Following Chronic Sympathetic Stimulation in The Aging Heart. East Tennessee State University;
- Ghosh D., Quach N., Pena C., Xuan B., Lee A., Dawson M. (2019). Mesenchymal Stem Cell Aging and Senescence Associated Extracellular Matrix Contributions to Breast Cancer Progression. Gordon Research Conference: Physical Science of Cancer;
- May J. Reed, Mamatha Damodarasamy, William A. Banks. (2019). The extracellular matrix of the blood–brain barrier: structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers. 7, 1651157;
- Juciano Gasparotto, Carolina S. Girardi, Nauana Somensi, Camila T. Ribeiro, José C.F. Moreira, et. al.. (2018). Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. Journal of Biological Chemistry. 293, 226-244;
- Shamim Shaikh, Louise F.B. Nicholson. (2008). Advanced glycation end products induce in vitro cross‐linking of α‐synuclein and accelerate the process of intracellular inclusion body formation. J. Neurosci. Res.. 86, 2071-2082;
- E. Schleicher, O.H. Wieland. (1986). Kinetic analysis of glycation as a tool for assessing the half-life of proteins. Biochimica et Biophysica Acta (BBA) - General Subjects. 884, 199-205;
- Ghulam Abbas, Ahmed Sulaiman Al-Harrasi, Hidayat Hussain, Javid Hussain, Rehana Rashid, M. Iqbal Choudhary. (2016). Antiglycation therapy: Discovery of promising antiglycation agents for the management of diabetic complications. Pharmaceutical Biology. 54, 198-206;
- Xu Wang, Song Yu, Chun-Yan Wang, Yue Wang, Hai-Xing Liu, et. al.. (2015). Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell.Dev.Biol.-Animal. 51, 204-209;
- H.-J. Luth. (2004). Age- and Stage-dependent Accumulation of Advanced Glycation End Products in Intracellular Deposits in Normal and Alzheimer's Disease Brains. Cerebral Cortex. 15, 211-220;
- Michael G. Friedrich, Zhen Wang, Kevin L. Schey, Roger J. W. Truscott. (2019). Mechanism of protein cleavage at asparagine leading to protein–protein cross-links. Biochemical Journal. 476, 3817-3834;
- Fülöp T., Dupuis G., Witkowski J.M., Larbi A. (2016). The Role of Immunosenescence in the Development of Age-Related Diseases. Revista de investigacion clinica; organo del Hospital de Enfermedades de la Nutricion. 68, 84–91;
- Valentin P. Yakubenko, Kui Cui, Christopher L. Ardell, Kathleen E. Brown, Xiaoxia Z. West, et. al.. (2018). Oxidative modifications of extracellular matrix promote the second wave of inflammation via β2 integrins. Blood. 132, 78-88;
- Sho-ichi Yamagishi, Kensei Taguchi, Kei Fukami. (2016). DNA-aptamers raised against AGEs as a blocker of various aging-related disorders. Glycoconj J. 33, 683-690;
- H. Chilukuri, M. J. Kulkarni, M. Fernandes. (2018). Revisiting amino acids and peptides as anti-glycation agents. Med. Chem. Commun.. 9, 614-624;
- Caroline Beaudoin-Chabot, Lei Wang, Alexey V. Smarun, Dragoslav Vidović, Mikhail S. Shchepinov, Guillaume Thibault. (2019). Deuterated Polyunsaturated Fatty Acids Reduce Oxidative Stress and Extend the Lifespan of C. elegans. Front. Physiol.. 10;
- Ji-young Lee, Jun-Gu Oh, Jin Sook Kim, Kwang-Won Lee. (2014). Effects of Chebulic Acid on Advanced Glycation Endproducts-Induced Collagen Cross-Links. Biol. Pharm. Bull.. 37, 1162-1167;
- Mohammad Nazrul Islam Bhuiyan, Shinya Mitsuhashi, Kengo Sigetomi, Makoto Ubukata. (2017). Quercetin inhibits advanced glycation end product formation via chelating metal ions, trapping methylglyoxal, and trapping reactive oxygen species. Bioscience, Biotechnology, and Biochemistry. 81, 882-890;
- Laurence Kennedy, Maria Pilar Solano, Luigi Meneghini, Margaret Lo, Margo P. Cohen. (2010). Anti-Glycation and Anti-Albuminuric Effects of GLY-230 in Human Diabetes. Am J Nephrol. 31, 110-116;
- David R. Sell, Klaus M. Biemel, Oliver Reihl, Markus O. Lederer, Christopher M. Strauch, Vincent M. Monnier. (2005). Glucosepane Is a Major Protein Cross-link of the Senescent Human Extracellular Matrix. Journal of Biological Chemistry. 280, 12310-12315;
- Matthew D. Streeter, Sheldon Rowan, Jason Ray, David M. McDonald, Jonathan Volkin, et. al.. (2020). Generation and Characterization of Anti-Glucosepane Antibodies Enabling Direct Detection of Glucosepane in Retinal Tissue. ACS Chem. Biol.. 15, 2655-2661;
- Vincent M. Monnier, David R. Sell. (2006). Prevention and Repair of Protein Damage by the Maillard Reaction In Vivo. Rejuvenation Research. 9, 264-273;
- Kazuhiko Ono, Manabu Nakashima. (2020). Sodium 4‑phenylbutyrate inhibits protein glycation. Biomed Rep. 13, 1-1;
- Nam Y. Kim, Tyler N. Goddard, Seungjung Sohn, David A. Spiegel, Jason M. Crawford. (2019). Biocatalytic Reversal of Advanced Glycation End Product Modification. ChemBioChem. 20, 2402-2410;
- Hye Eun Yoon, Bum Soon Choi. (2014). The renin-angiotensin system and aging in the kidney. Korean J Intern Med. 29, 291;
- Keisuke Okuno, Stephanie Cicalese, Katherine J. Elliott, Tatsuo Kawai, Tomoki Hashimoto, Satoru Eguchi. (2020). Targeting Molecular Mechanism of Vascular Smooth Muscle Senescence Induced by Angiotensin II, A Potential Therapy via Senolytics and Senomorphics. IJMS. 21, 6579;
- Sara Conti, Paola Cassis, Ariela Benigni. (2012). Aging and the Renin-Angiotensin System. Hypertension. 60, 878-883;
- Aurelie Nguyen Dinh Cat, Augusto C. Montezano, Dylan Burger, Rhian M. Touyz. (2013). Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxidants & Redox Signaling. 19, 1110-1120;
- Wang C.H., Li F., Takahashi N. (2010). The renin angiotensin system and the metabolic syndrome. The open hypertension journal. 3, 1–13;
- Keisuke Okuno, Stephanie Cicalese, Katherine J. Elliott, Tatsuo Kawai, Tomoki Hashimoto, Satoru Eguchi. (2020). Targeting Molecular Mechanism of Vascular Smooth Muscle Senescence Induced by Angiotensin II, A Potential Therapy via Senolytics and Senomorphics. IJMS. 21, 6579;
- Alexander Fedintsev, Alexey Moskalev. (2020). Stochastic non-enzymatic modification of long-lived macromolecules - A missing hallmark of aging. Ageing Research Reviews. 62, 101097;
- Kim S. and Iwao H. (2000). Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacological reviews. 1, 11–34;
- Hiroaki Kawano, Yung S. Do, Yasuko Kawano, Vaughn Starnes, Mark Barr, et. al.. (2000). Angiotensin II Has Multiple Profibrotic Effects in Human Cardiac Fibroblasts. Circulation. 101, 1130-1137;
- Andrea I. Alford, Andrew Z. Golicz, Amber Lee Cathey, Anita B. Reddy. (2013). Thrombospondin-2 facilitates assembly of a type-I collagen-rich matrix in marrow stromal cells undergoing osteoblastic differentiation. Connective Tissue Research. 54, 275-282;
- C Kupfahl. (2000). Angiotensin II directly increases transforming growth factor β1 and osteopontin and indirectly affects collagen mRNA expression in the human heart. Cardiovascular Research. 46, 463-475;
- Sergio A. Mezzano, Marta Ruiz-Ortega, Jesús Egido. (2001). Angiotensin II and Renal Fibrosis. Hypertension. 38, 635-638;
- Franck Verrecchia, Alain Mauviel. (2007). Transforming growth factor-β and fibrosis. WJG. 13, 3056;
- Andreea Iren Serban, Loredana Stanca, Ovidiu Ionut Geicu, Maria Cristina Munteanu, Anca Dinischiotu. (2016). RAGE and TGF-β1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS ONE. 11, e0152376;
- Aristidis Moustakas, Carl-Henrik Heldin. (2008). Dynamic control of TGF-β signaling and its links to the cytoskeleton. FEBS Letters. 582, 2051-2065;
- Young-Sil Yoon, Jae-Ho Lee, Sung-Chul Hwang, Kyeong Sook Choi, Gyesoon Yoon. (2005). TGF β1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 24, 1895-1903;
- Lina Vardouli, Eleftheria Vasilaki, Elsa Papadimitriou, Dimitris Kardassis, Christos Stournaras. (2008). A novel mechanism of TGFβ-induced actin reorganization mediated by Smad proteins and Rho GTPases. FEBS Journal. 275, 4074-4087;
- Elena MV de Cavanagh, Marcelo Ferder, Felipe Inserra, Leon Ferder. (2009). Angiotensin II, mitochondria, cytoskeletal, and extracellular matrix connections: an integrating viewpoint. American Journal of Physiology-Heart and Circulatory Physiology. 296, H550-H558;
- Mitch Leslie. (2011). Stiff ECM puts the brakes on microtubule growth. Journal of Cell Biology. 192, 204-204;
- Lee A. Ligon, Oswald Steward. (2000). Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol.. 427, 351-361;
- Thomas H. Ambrosi, Antonio Scialdone, Antonia Graja, Sabrina Gohlke, Anne-Marie Jank, et. al.. (2017). Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell. 20, 771-784.e6;
- Natasha Baker, Lisa B. Boyette, Rocky S. Tuan. (2015). Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone. 70, 37-47;
- Laëtitia Gorisse, Christine Pietrement, Vincent Vuiblet, Christian E. H. Schmelzer, Martin Köhler, et. al.. (2016). Protein carbamylation is a hallmark of aging. Proc Natl Acad Sci USA. 113, 1191-1196;
- Matthew D. Streeter, Sheldon Rowan, Jason Ray, David M. McDonald, Jonathan Volkin, et. al.. (2020). Generation and Characterization of Anti-Glucosepane Antibodies Enabling Direct Detection of Glucosepane in Retinal Tissue. ACS Chem. Biol.. 15, 2655-2661;
- Altaf S Darvesh, Richard T Carroll, Anupam Bishayee, Werner J Geldenhuys, Cornelis J Van der Schyf. (2010). Oxidative stress and Alzheimer’s disease: dietary polyphenols as potential therapeutic agents. Expert Review of Neurotherapeutics. 10, 729-745;
- Hideaki Kaneto, Naoto Katakami, Munehide Matsuhisa, Taka-aki Matsuoka. (2010). Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis. Mediators of Inflammation. 2010, 1-11;
- Romano A. D., Serviddio G., de Matthaeis A., Bellanti F., Vendemiale G. (2010). Oxidative stress and aging. Journal of nephrology. 23 Suppl 15, S29–S36;
- Emanuele Marzetti, Riccardo Calvani, Matteo Cesari, Thomas W. Buford, Maria Lorenzi, et. al.. (2013). Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. The International Journal of Biochemistry & Cell Biology. 45, 2288-2301;
- Stephen E. Alway, Junaith S. Mohamed, Matthew J. Myers. (2017). Mitochondria Initiate and Regulate Sarcopenia. Exercise and Sport Sciences Reviews. 45, 58-69;
- Denise Zdzieblik, Steffen Oesser, Manfred W. Baumstark, Albert Gollhofer, Daniel König. (2015). Collagen peptide supplementation in combination with resistance training improves body composition and increases muscle strength in elderly sarcopenic men: a randomised controlled trial. Br J Nutr. 114, 1237-1245;
- Luc E. Gosselin. (2011). Skeletal Muscle Collagen: Age, Injury and Disease. Sarcopenia – Age-Related Muscle Wasting and Weakness. 159-172;
- Masahiro Tada, Yutaro Yamada, Koji Mandai, Noriaki Hidaka. (2018). Matrix metalloprotease 3 is associated with sarcopenia in rheumatoid arthritis ‐ results from the CHIKARA study. Int J Rheum Dis. 21, 1962-1969;
- Aicha Melouane, Antoine Carbonell, Mayumi Yoshioka, Jack Puymirat, Jonny St-Amand. (2018). Implication of SPARC in the modulation of the extracellular matrix and mitochondrial function in muscle cells. PLoS ONE. 13, e0192714;
- Isabella T. Tai, Michelle J. Tang. (2008). SPARC in cancer biology: Its role in cancer progression and potential for therapy. Drug Resistance Updates. 11, 231-246;
- H Sage, R B Vernon, S E Funk, E A Everitt, J Angello. (1989). SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2-dependent binding to the extracellular matrix.. Journal of Cell Biology. 109, 341-356;
- Haiyan Song, Yuanyuan Guan, Liping Zhang, Kun Li, Chunli Dong. (2010). SPARC interacts with AMPK and regulates GLUT4 expression. Biochemical and Biophysical Research Communications. 396, 961-966;
- Aicha Melouane, Mayumi Yoshioka, Jonny St-Amand. (2020). Extracellular matrix/mitochondria pathway: A novel potential target for sarcopenia. Mitochondrion. 50, 63-70;
- Tomoyasu Kadoguchi, Shintaro Kinugawa, Shingo Takada, Arata Fukushima, Takaaki Furihata, et. al.. (2015). Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol. 100, 312-322;
- Alexander Fedintsev, Alexey Moskalev. (2020). Stochastic non-enzymatic modification of long-lived macromolecules - A missing hallmark of aging. Ageing Research Reviews. 62, 101097;
- Hewitt J. (2013). Protein lifetime and the stability of cell structures. Phys.org;
- Sajithlal G.B. and Chandrakasan G. (1999). Role of lipid peroxidation products in the formation of advanced glycation end products: An in vitro study on collagen. Proc. Indian Acad. Sci. (Chem. Sci.). 111, 215–229;
- Noemi A. Zambetti, Zhen Ping, Si Chen, Keane J.G. Kenswil, Maria A. Mylona, et. al.. (2016). Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell. 19, 613-627.
Комментарии
Раньше здесь был блок с комментариями. Но потом сервис Disqus, на котором они работали и за который мы платили, перестал открываться из РФ.
Когда появится возможность, мы вернём комментарии уже на внутреннем движке, а чтобы это произошло быстрее —
Оставьте донат 💚

