На заре молекулярной графики
15 марта 2007
На заре молекулярной графики
- 8683
- 0
- 9

Изображение Лайнуса Полинга и альфа-спирали маслом. Автор — Леон Тадрик (© 1951).
Развитие биологии в XX веке неразрывно соединилось с изучением молекулярных основ жизни. Бурный прогресс биохимии, биофизики и молекулярной биологии привел к тому, что для многих важнейших процессов были установлены определяющие их молекулярные механизмы. В связи с этим появилась насущная потребность в визуализации молекул, способной дать представление об их пространственной организации, и, следовательно, дать ключ к объяснению их функций. В данном обзоре рассмотрены основные этапы развития визуальных способов представления структуры биомакромолекул — от «физических» моделей до первых компьютерных алгоритмов молекулярной графики.
Введение
Издавна информацию о составе и строении химического вещества принято было записывать с помощью химических формул, по которым можно судить об элементном составе вещества и его молекулярной массе. Однако такая простейшая форма записи часто не может дать понятия о строении молекулы (как, например, «эмпирическая» формула гемоглобина — C3032Н4816N780О872N780S8Fe4 — практически ничего не говорит о его устройстве). Для сложных веществ, обладающих высокой конформационной подвижностью, даже структурная формула (графическое изображение связанных между собой химических групп на плоскости) не позволяет описать пространственное строение молекулы.
В 40-х годах прошлого века началось бурное развитие структурной химии и рентгеновской кристаллографии, что привело к определению первых пространственных структур органических молекул. Возможности структурных формул оказались исчерпаны, так как с их помощью (хоть они и называются структурными) крайне затруднительно передать трехмерное строение сложной молекулы. Вскоре последовавшие открытия структур белковых молекул и ДНК создали дополнительную потребность в способах наглядного изображения пространственной структуры биомакромолекул.
В течение третьей четверти XX века структуру молекул передавали практически исключительно с помощью так называемых «физических моделей» (см. главу «“Физические” модели макромолекул»), то есть буквально «собирали» молекулу в увеличенном масштабе с использованием подручных материалов: пластмассовых шариков, медных стерженьков и тому подобного. С развитием вычислительной техники визуализация молекул стала проводиться почти полностью на дисплеях компьютеров, однако построение «физических» моделей не кануло в лету: они по-прежнему используются в качестве учебных пособий и служат источником вдохновения для людей, занимающихся молекулярной скульптурой (см. главу «“Молекулярная” скульптура»).
«Физические» модели макромолекул
Самые знаменитые модели молекул: белковая α-спираль и двойная спираль ДНК
Середина XX века была ознаменована двумя крупнейшими открытиями, положившими начало структурной биологии и невероятно продвинувшими вперед все биологические науки. Эти открытия связаны с установлением принципов пространственной организации двух классов биологически важных макромолекул: белков и дезоксирибонуклеиновой кислоты (ДНК).
Одним из основателей структурной биологии по праву считается Лайнус Полинг (Linus Carl Pauling) [1], след которого в науке и других сферах деятельности чрезвычайно глубок: это и развитие рентгеновской кристаллографии, и создание теории химического резонанса, и объяснение молекулярных основ серповидной анемии, и активная антимилитаристическая деятельность (отмеченная в 1962 году нобелевской премией Мира). Одно из наиболее существенных его достижений (также отмеченное нобелевской премией (рис. 1)) основывается на изучении структурных особенностей фибриллярных белков. Совместно с Робертом Кори (Robert Corey) Полинг открыл один из самых распространённых мотивов укладки полипептидной цепи — α-спираль. (Их открытие, обнародованное в 1951-м году после 14 лет упорного труда [2], не связано с детальным описанием структуры какого-либо белка: это было сделано семью годами позже (см. главу «Каркасные (“проволочные”) модели»).
Лайнус Полинг был очень экспансивным, энергичным и нетерпеливым человеком: говорят, что своё открытие он сделал, руководствуясь более «химической интуицией» и молекулярными моделями полипептидной цепи, чем данными рентгеновской кристаллографии и расчётами длин химических связей, выполненными его более усидчивым коллегой Р. Кори. Для определения возможных конфигураций белковой цепи Полинг строил разнообразные модели, с помощью которых можно было установить, подчиняется ли эта конфигурация стереохимическим закономерностям, в частности, установленному им факту, что пептидная связь (состоящая из четырёх атомов) должна быть плоской.
Модели атомов и аминокислотных остатков должны «автоматически» (посредством своей конструкции) учитывать такие характеристики, как длины и углы валентных связей, возможные значения двугранных углов, способность некоторых пар атомов образовывать водородные связи и пр. Модели, используемые Полингом и Кори (рис. 1), позволили им сделать вывод, что главная цепь белковой молекулы может укладываться в спиральную структуру с нецелым числом аминокислотных остатков (3,6 остатка), приходящихся на один виток спирали. При этом спираль стабилизируется водородными связями между атомами главной цепи остатков, смещённых друг относительно друга на четыре позиции. Полученная в 1958 году пространственная структура миоглобина (см. главу «Каркасные (“проволочные”) модели») подтвердила сделанное Полингом открытие и показала, что α-спираль — один из самых широко распространённых элементов конструкции белковой молекулы.

Рисунок 1. Слева: Лайнус Полинг и Роберт Кори с моделью α-спирали (1951). Справа: Лайнус Полинг держит в руках модель молекулы сульфаниламида (1954). Атомы в модели Полинга были изготовлены из дерева (в масштабе 1’’/Å) или пластика (0,5’’/Å). В 1954 году «за изучение природы химической связи и его применение к объяснению строения сложных молекул» Полинг был удостоен Нобелевской премии по химии [1], [4].
сайт «Лайнус Полинг и гонка за ДНК»
Лайнус Полинг рассказывает о том, как он открыл α-спираль.
Однажды, когда я работал в Оксфорде (дело было в апреле 1948 года), я заболел, и после двух дней в постели, проведенных за чтением фантастики и детективов, я подумал: „А не открыть ли мне α-спираль?“, или что-то вроде этого. Чтобы понять, как устроена полипептидная цепь с учётом всех требований структурной химии и как водородные связи поддерживают структуру белка, я взял лист бумаги навроде этого [берет лист бумаги] и нарисовал на нем полипептидную цепь в распрямлённой конформации (кстати, этот лист остался у меня до сих пор). Я стал складывать лист бумаги так, чтобы угол между Cα-атомами соседних остатков составлял около 110° [складывает листок], и после нескольких попыток сложил его таким образом, чтобы каждая NH и CO группа участвовала в образовании связи N-H···O=C. Я понял, что эта структура, присутствующая в белках, выделенных из волос, рога, ногтей и мышечных волокон, называемая α-спиралью, содержит 3,6 аминокислотных остатка на один виток спирали.
Джеймс Уотсон (James Watson) и Френсис Крик (Francis Crick), первооткрыватели структуры ДНК в форме двух комплементарных цепей, образующих двойную спираль, в своей работе также руководствовались молекулярными моделями. Их коллеги, занимавшиеся дифракцией рентгеновских лучей на образцах ДНК, — Морис Уилкинс (Maurice Wilkins) и Розалинд Фрэнклин (Rosalind Franklin) — несколько скептически относились к затее с моделями, полагая, что строение ДНК может быть определено исключительно из эксперимента. Однако именно «физические» модели вместе с гениальной догадкой о «комплементарности» цепей ДНК (основанной на «правилах Чаргаффа») позволили свершиться этому важнейшему открытию.
Уотсон, видимо, был настолько поражён элегантным открытием Полинга и его моделями, что в своей работе делал ставку главным образом на молекулярные модели как инструмент, с помощью которого можно было объяснить дифракционные данные, полученные Фрэнклин:
Однако в ближайшие несколько дней ни одной серьезной модели мы не построили. Нам не только не хватало моделей пуриновых и пиримидиновых оснований, но мастерская так и не изготовила для нас ни одной модели атома фосфора. Для того чтобы сделать даже самые простые атомы фосфора, нашему механику требовалось не менее трех дней, а потому после обеда я пошел к себе в Клэр-колледж привести в порядок статью по генетике.
Фрэнсис все больше тревожился из-за того, что я перестал работать над молекулярными моделями. … Чуть ли не каждый день после обеденного перерыва он то и дело раздраженно косился на заброшенный полинуклеотидный остов, зная, что я тем временем играю где-нибудь в теннис. … Брюзжание Фрэнсиса меня не беспокоило — усовершенствовать дальше наш последний остов не имело смысла, пока не будет решена проблема оснований.
Составные части моделей, с которыми работали Уотсон и Крик, были изготовлены в мастерских Кавендишской лаборатории по их собственным чертежам и схемам. Их модели были устроены иначе, чем модели Полинга: если у последнего атомы были выполнены в масштабе и заполняли пространство довольно плотно (space filling), то Уотсон и Крик работали с «проволочными» моделями, в которых атомы сравнительно небольшого радиуса были соединены отрезками медной проволоки, изображавшими химические связи (рис. 2).
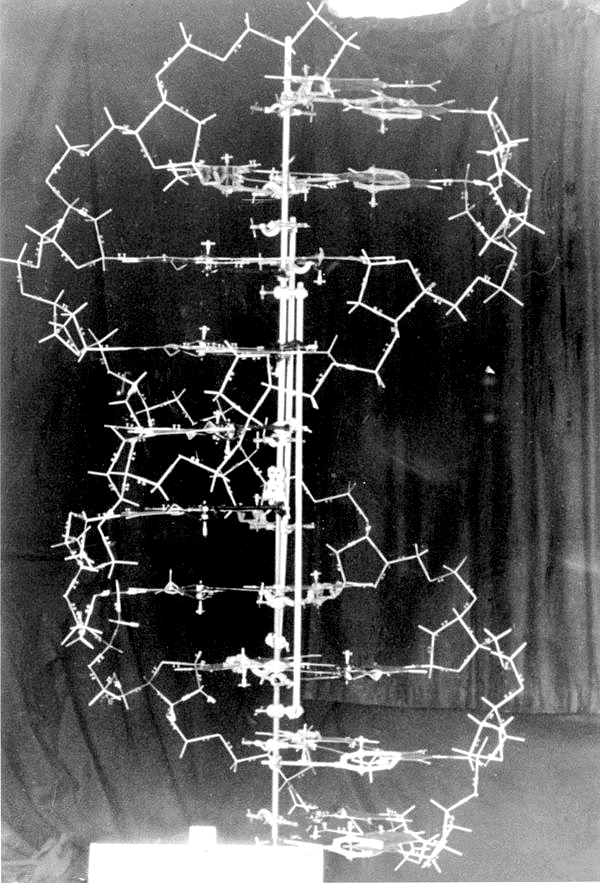
Рисунок 2а. Оригинальная модель двойной спирали ДНК, построенная Уотсоном и Криком. Увлекательная история об открытии этой структуры и роли других людей в данном исследовании (а также о том, как Полинг чуть не опередил их в открытии структуры ДНК, но пошёл ложным путём, решив, что молекула организована в виде тройной спирали) подробно описаны в книге Уотсона «Двойная спираль» [3].
архив лаборатории Колд Спринг Харбор

Рисунок 2б. Джеймс Уотсон с моделью ДНК (1957). В 1962 году совместно с Френсисом Криком и Моррисом Уилкинсом он был награждён Нобелевской премией по медицине за «открытия в области молекулярной структуры нуклеиновых кислот и их значимости для передачи генетической информации» [5]. К сожалению, Розалинд Фрэнклин, полноправный участник этого открытия, умерла от рака в 1958 году, а правила Нобелевского комитета не позволяют вручать премию посмертно.
Френсис Крик рассказывает об открытии структуры ДНК в виде двух антипараллельных комплементарных друг другу цепей, образующих двойную спираль, и демонстрирует молекулярную модель ДНК.
Модель Полинга оказалась очень удачной, и на её основе был разработан промышленный стандарт и выпущены многочисленные наборы для построения моделей молекул.
Модель Кори–Полинга–Колтуна: стандарт «молекулярного конструктора»
После того, как общественность осознала значение открытий, сделанных Полингом и Уотсоном, польза молекулярных моделей стала очевидной. Встала задача промышленного выпуска наборов, позволяющих без особых усилий собирать модели молекул, поскольку предыдущее их поколение было ненадёжным, некачественным и дорогим в изготовлении. В мае 1960-го года комитет по биофизике и биофизической химии (Biophysics and Biophysical Chemistry Study Section) Национального Института Здравоохранения (НИЗ) США обратился к Уолтеру Колтуну (Walter Koltun) с просьбой создать промышленные спецификации для выпуска «молекулярного конструктора», основываясь на моделях, разработанных Полингом и Кори. Обоснование этой программы, рассчитанной на пять лет, было следующим:
«Модели атомов, доступные в настоящее время, обладают рядом недостатков. Некоторые из них основаны на металлических клипсах, самопроизвольно расстегивающихся при построении больших моделей. Они практически не обеспечивают подвижность молекулы и не позволяют варьировать углы и длины химических связей. Торсионный потенциал и способность атомов к образованию водородных связей также не учитываются. Кроме того, модели атомов, как правило, настолько тяжелы, что соединительные элементы не способны удерживать их в правильной конформации. Лёгкие же модели обладают недостаточной точностью. В добавок ко всему, эти модели дороги в изготовлении и стоят в среднем 75 центов за атом» [6].
В результате были созданы модели, учитывающие требования комитета (рис. 3). Они получили название «Новые модели атомов по Кори–Полингу с соединительными элементами Колтуна», или, для краткости, CPK-модели (Corey–Pauling–Koltun). Модели оказались настолько удачными, что используются до сих пор, а вид компьютерного изображения молекулы с атомами в виде сфер в масштабе ван-дер-ваальсовых радиусов атомов так и называется: CPK. Тогда же и была принята схема цветового обозначения атомов, популярная и по сей день: C — черный, N — синий, H — белый, O — красный.

Рисунок 3. Слева: Фрагмент рисунка из патента, выданного Колтуну на его изобретение (1962). Рисунок объясняет устройство соединительных элементов, используемых для сборки моделей. Модели атомов и соединительные элементы были спроектированы с учётом требований, выдвинутых комитетом разработчику (см. ниже). Справа: Модель молекулы дипептида ГЛУ–ТРП, собранная из CPK-конструктора, найденного в лаборатории моделирования биомолекулярных систем Института биоорганической химии РАН (краешек пальца на фотографии мой. — А. Ч.).
Характеристики молекулярных моделей C-P-K [6]:
- Размеры. Углы и длины связей, а также ван-дер-ваальсовы радиусы атомов должны соответствовать наиболее актуальным научным данным. Валентные углы должны воспроизводиться с допуском ±0°30’, длины связей — ±0,01 Å, а радиусы атомов — ±0,03 Å;
- Масштаб должен составлять 1,25 см/Å. Этого достаточно для построения точных моделей при невысокой стоимости изготовления. Так, молекула с размерами 20×40×100 Å будет изображена моделью 25×50×125 см;
- Вес моделей атомов должен быть настолько мал, насколько это возможно (без нарушения остальных требований);
- Ограничение на вращение. Связи, не способные к вращению, должны быть зафиксированы. В остальных случаях вращение должно быть ограничено в соответствии с торсионным потенциалом;
- Водородные связи. В моделях должна присутствовать возможность образования водородных связей, столь существенных для функций макромолекул;
- Типы атомов. Разнообразие типов атомов в наборе должно позволять конструкцию всех практически важных типов биологических молекул;
- Спецатомы. Должна быть предусмотрена возможность изготовления спецатомов, важных в ряде случаев (хоть и не выпускаемых массово из-за редкого использования). Эти атомы должны быть совместимы с другими атомами в наборе;
- Соединительные элементы должны (а) надёжно фиксировать атомы; (б) позволять варьирование валентных углов в диапазоне ±8° без существенных усилий; (в) предотвращать самопроизвольное проворачивание групп атомов (до 50) под действием их собственного веса и не прогибаться более чем на 3° под этим весом и (г) позволять варьирование длин связей (в разумных пределах);
- Материалы и стоимость. Модели должны быть изготовлены из пластмассы со средней стоимостью около 15 центов за атом. Эта цена учитывает массовое производство и возможность государственной субсидии на изготовление сложных форм для отливки.
Каркасные («проволочные») модели
Первая пространственная структура белка, полученная Джоном Кендрю (John Kendrew) в 1958 году [7] с помощью анализа дифракции рентгеновских лучей на белковом кристалле, поразила учёных в первую очередь тем, что в ней не наблюдалось никакой симметрии. Многочисленные α-спиральные элементы в структуре миоглобина (именно этот белок исследовал Кендрю) располагались в пространстве прихотливым образом, подтверждая открытие Полинга о том, что α-спираль — один из основных типов укладки полипептидной цепи.
Первоначально полученная с пространственным разрешением 6 Å, модель позволяла различить лишь основную укладку белка, но не точное расположение боковых цепей аминокислотных остатков (рис. 4А). Дальнейшая оптимизация рентгенографической процедуры и применение вычислительной техники позволили поднять разрешение до 2 Å, и в результате была построена каркасная (или «проволочная») модель миоглобина в масштабе 5 см/Å. Модель, построенная из медных трубок, изготовленных в мастерских Кембриджа, располагалась внутри полого куба с ребром 2 м и поддерживалась ~2500 вертикальными металлическими палочками, затруднявшими её построение, восприятие и переноску (рис. 4В).
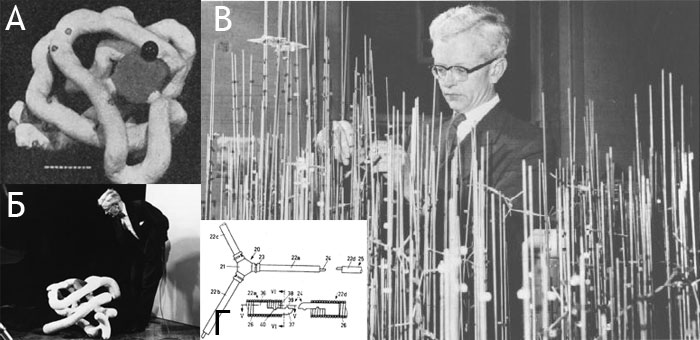
Рисунок 4. Модель молекулы миоглобина, построенная Кендрю и сотр. А. Фотография модели электронной плотности миоглобина, вылепленной из пластилина [7]. Белым цветом показана полипептидная цепь, серый диск соответствует гемовой группировке, сферами обозначены атомы тяжёлых металлов, использованные в процедуре изоморфного замещения. Короткий штрих на масштабной линейке равен 1 Å. Б. Кендрю демонстрирует модель миоглобина. В. Кендрю за изготовлением своей «проволочной» модели. Многочисленные вертикальные металлические палочки поддерживают полипептидную цепь. Г. Фрагмент изображения из патента, полученного Андре Дрейдингом (Andre Dreiding) на конструкцию каркасной молекулярной модели из нержавеющей стали.
Модель, построенная Кендрю, была очень сложна в обращении и почти не поддавалась модификации, и, когда потребовалось построить несколько копий этой модели по запросу других университетов и лабораторий, возникла проблема создания более надёжной и удобной конструкции. В решении этой задачи помог Баркер (A.A. Barker), сотрудник инженерной лаборатории в Кембридже. Начиная с 1965 года, он конструировал молекулярные модели различных биомолекул, включая ДНК, витамин B12, инсулин, α-спираль и др. Кендрю предложил Баркеру использовать компоненты, изготовленные Биверсом (C.A. Beevers), профессором химии из Эдинбургского университета, — небольшие пластиковые шарики диаметром 6.9 мм с просверленными в них с помощью специального устройства отверстиями, в которые вставлялись лёгкие металлические трубочки. В первые же годы (с 1966 по 1968) Кендрю поступило около 30 заказов на молекулярные модели миоглобина и других белков, изготавливаемые в масштабе 1 см/Å примерно за месяц на одну модель (рис. 5). Цена моделей была около 600$, что являлось весьма существенной суммой в те времена.
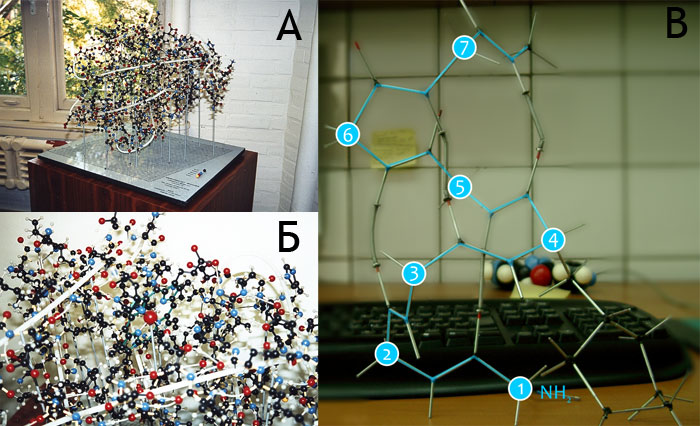
Рисунок 5. А, Б. Каркасные модели молекулы миоглобина, построенные Баркером. Главная цепь белка изображена белым шнуром. На подставке рядом с моделью (А) расположена «легенда» с расшифровкой использованных цветов и размеров атомов. Данный экземпляр модели принадлежит Бриттону Чансу (Britton Chance). В. Небольшой участок α-спирали, построенный из «проволочного» конструктора, найденного в лаборатории моделирования биомолекулярных систем. Для упрощения восприятия, ход главной цепи «подсвечен» голубой линией, а остатки пронумерованы по Cα-атомам. Водородные связи создаются с помощью специальных пружинок. (У четвертого остатка присутствует боковая цепь, построенная из «атомов» углерода в sp3 гибридизации, поскольку других найдено не было, также как и отдельных моделей боковых цепей. Так что это не фенилаланин. — А.Ч.)
Модели, изображающие электронную плотность
Хотя первая пространственная структура белка была получена Джоном Кендрю, истинным первооткрывателем в данной области является другой человек — Макс Перутц (Max Perutz). Именно ему принадлежит идея изоморфного замещения, когда в определённые позиции белковой молекулы вводится атом тяжёлого металла, и на основании получаемых при этом искажений дифракционной картины появляется возможность установить структуру молекулы. В 1960 году Перутцем и его коллегами была получена пространственная структура гемоглобина [8] — белка существенно более сложного, нежели миоглобин.
Для визуализации пространственных карт электронной плотности Перутц использовал более количественный метод, чем Кендрю в своей первой структуре. Изопотенциальные области электронной плотности выпиливались из толстого куска пластмассы и скреплялись стопками, что позволило изобразить пространственную структуру молекулы (рис. 6).

Рисунок 6. Слева: Модель электронной плотности β-субъединицы оксигемоглобина лошади [9] с пронумерованными α-спиральными элементами. В верхнем углу приведены «идеализированные» β-цепи гемоглобина (также показана ось двукратной симметрии). Две α-субьединицы, образующие аналогичную пару, тут не показаны. Справа: Макс Перутц с полной (тетрамерной) моделью электронной плотности молекулы гемоглобина и Джон Кендрю, каркасная модель молекулы миоглобина которого стоит на переднем плане. Фотография сделана в 1962 году на вручении Нобелевской премии по химии, которую Перутц с Кендрю получили «за исследования структуры глобулярных белков» [10].
Макс Перутц рассказывает историю открытия структуры гемоглобина. Интервью было записано в 2001 году, незадолго до его смерти (Макс Перутц умер 6 февраля 2002 года).
Дальнейшая автоматизация построения моделей и «вписывания» их в трехмерные карты электронных плотностей была произведена Фредом Ричардсом (Fred Richards) и его коллегами. С помощью сконструированного ими оптического устройства, названного «коробкой Ричардса» (Richards box), двумерные изопотенциальные карты электронной плотности, построенные на кальке с помощью компьютера, переносились на прозрачные подложки и закреплялись с равными промежутками внутри «коробки». Полученные таким образом трёхмерные карты строились в том же масштабе, что и молекулярная модель (обычно 2 см/Å), и с помощью системы полупрозрачных зеркал оптически совмещались с моделью, причём «качество» совмещения можно было оценить немедля, без длительного измерения углов и расстояний (рис. 7). «Коробка Ричардса» была использована при расшифровке структуры рибонуклеазы [11], и до конца 1970-х оставалась стандартом де-факто при определении структур белков с помощью рентгеновской кристаллографии.
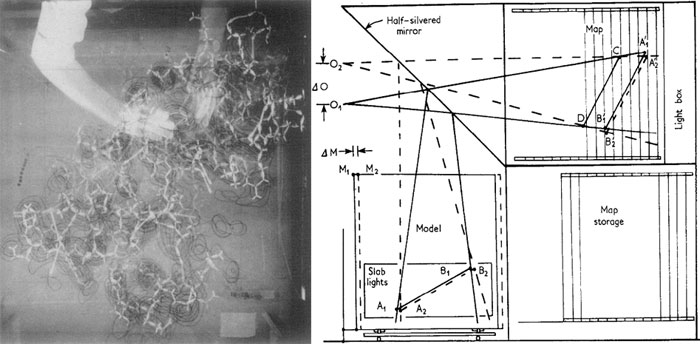
Рисунок 7. «Коробка Ричардса». Слева: Фотография, сделанная в процессе ручного «вписывания» модели рибонуклеазы-S в трехмерные карты электронной плотности [11] (устройство снимало стереоизображение; только одна из двух фотографий приведена здесь). Исследователь (видимо, Ричардс) занимается подстраиванием геометрии остатка ФЕН46 для лучшего соответствия карте электронной плотности. Справа: Схематическое устройство «коробки».
Другие модели полипептидной цепи
Молекулярные модели, описанные выше, были очень громоздки, тяжелы и сложны в изготовлении. Байрон Рубин (Byron Rubin) в начале 1970-х построил устройство, которое могло автоматически строить упрощённые модели белковых молекул (только контуры главной цепи по Cα-атомам, рис. 8А), изгибая толстую проволоку согласно заложенной в него программе. Модели, построенные на «сгибателе Байрона» (именно так назвали машину), были весьма компактны, легки и очень наглядны (хотя и, понятно, не могли обеспечить того уровня подробности, как модели, включающие атомы боковых цепей). Важность наглядного представления молекулы белка (пусть и в упрощённой форме) вскоре была подтверждена на практике. На одну из научных конференций в середине 70-х, когда уже было известно более десятка белковых структур, Дэвид Дэвис (David Davies) привёз модель антиген-связывающего домена иммуноглобулина (Fab-фрагмент), а Джейн и Дэвид Ричардсон (Jane и David Richardson) — модель супероксид-дисмутазы, построенные на сгибателе Байрона. Разглядывая модели, исследователи поняли, что их пространственное строение очень близко, несмотря на то, что идентичность аминокислотных последовательностей этих белков не превышает 9%. Так впервые был обнаружен иммуноглобулиновый домен, позже найденный во множестве других белков, непосредственно не связанных с иммунитетом.
Позже (в 1982 г.) была представлена Блэкуэлльская модель (Blackwell Molecular Models), также предназначенная для изображения конформации главной цепи белка, но существенно более сложная и точная, чем «сгибатель». Пластмассовые шарики двадцати разных цветов, соответствующие различным аминокислотам, были снабжены парой прецизионных шарниров, позволявших фиксировать двугранные и «валентные» углы, задающие конформацию полипептидной цепи (значения углов φ и ψ тут не определены, поскольку одному остатку соответствует только один «атом», рис. 8Б). Необходимые для построения модели расчёты координат «атомов» проводились на компьютере, оптимизируя геометрию молекулы с тем, чтобы длины «связей» в модели были равны 3.8 см. В это время были предложены и другие системы для построения «физических» молекулярных моделей (рис. 8В), но я на них подробно останавливаться не буду.
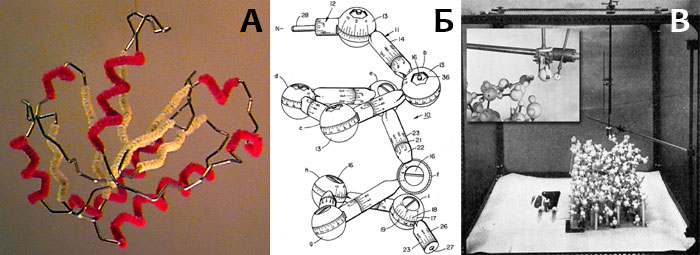
Рисунок 8. А. Каркасная модель основной цепи l-домена интегрина CD11a (код PDB: 1LFA), построенная на сгибателе Байрона. α-Спирали обёрнуты красным поролоном, β-листы — жёлтым. Сгибатель Байрона выпускался вплоть до конца 1990-х. Тим Герман (Tim Herman) — автор изображённой модели — стал одним из последних его пользователей, применяя аппарат на занятиях во многих университетах США. Б. Рисунок из патента, полученную на Блэкуэлльскую молекулярную модель. В. «Простое устройство» для автоматического конструирования сложных молекулярных моделей (A simple jig for building complex molecular models [14]).
С течением времени всё большее значение в визуализации биомакромолекул и построении кристаллографических моделей белков стали приобретать компьютеры, но конструкция «физических» моделей не канула в лету: кроме учебных применений, они стали объектом особой области искусства — «молекулярной» скульптуры.
«Молекулярная» скульптура
Вскоре после создания «сгибателя» Байрон Рубин адаптировал свою машину к «сырью» более крупного диаметра, и построил модель рубредоксина из труб, используемых для автомобильных глушителей. Он и по сей день занимается молекулярной скульптурой (рис. 9А, Б). Гроссман (Bathsheba Grossman) занимается лазерной гравировкой биомолекул в стеклянных блоках (рис. 9В, Г), Восс-Андре (Julian Voss-Andreae) создаёт модели молекул из металла (рис. 9Д), Мейер (Edgar Meyer) — из дерева. Эвард (Kenneth Eward) ведет виртуальный музей молекулярной скульптуры. Возле института Биоорганической Химии РАН в Москве стоит памятник комплексу валиномицина с ионом калия (рис. 9Е).

Рисунок 9. Молекулярная скульптура. А. Скульптура коллагеназы, выделенной из нейтрофилов человека, изваянная Байроном Рубином (© 2001) (выставлена около Смитсоновского института в штате Вашингтон, США). Высота около 30 см. Б. Байрон Рубин за работой. В, Г. Изображение молекулы ДНК, выгравированное с помощью лазера в стеклянном параллелепипеде (изготовлено Гроссманом). Д. Скульптура «Феромон», изготовленная из нержавеющей стали Восс-Андре (высота 90 см). Е. Скульптура «Валиномицин», установленная около входа в институт Биоорганической химии в Москве, «изображает комплекс антибиотика валиномицина с ионом калия». «Общий принцип связывания ионов металлов и их перенос через мембраны с помощью ионофоров был открыт в институте в 1963 году.» (Из надписи на табличке.)
В настоящее время при построении физических моделей молекул могут использоваться технологии трехмерного прототипирования — такие как «трехмерные» принтеры, слой за слоем создающие трехмерный объект по заданной компьютером программе. (Подробнее о молекулярной скульптуре можно почитать в «Компьютерре» [23])
Начало компьютерной эры в визуализации молекул
Первая система компьютерной визуализации
Уже в 1964 году Сайрус Левинталь (Cyrus Levinthal) и его коллеги в Массачусетском Технологическом Институте (МТИ) разработали электронную систему, изображавшую основную цепь белка на экране осциллографа с возможностью вращения этой модели с помощью специального трекбола. Расчёты, необходимые для вывода картинки на экран осциллографа (рис. 10), производились на одном из первых мейнфреймов (проект “Multi-Access Computer”).
Пока еще невозможно оценить все преимущества, которые может дать компьютер при решении современных проблем молекулярной биологии. Однако, очевидно, что комбинация человек-машина может быть чрезвычайно эффективной. Уже показано, что компьютер может использоваться для построения и визуализации крупных молекул, и это его применение весьма полезно для понимания механизмов функционирования молекул. В то же время, не один год пройдёт, прежде чем полностью будет осознано, насколько важно интерактивное общение с компьютером в процессе построения модели белка.

Рисунок 10. Система молекулярного моделирования Левинталя. Слева: Экран (электронно-лучевая трубка) осциллографа с выведенной на нём моделью белкового остова. Вращение молекулы на экране контролируется с помощью стоящей рядом полусферы-«трекбола». Справа: Общий вид системы. Фото Мартина Цвика (Martin Zwick).
Фильм «Белки» (1966), переснятый с экрана Левинталевской системы на 16-мм киноплёнку (эпизод «Миоглобин», показывающий структуру, полученную Кендрю (см. главу «Каркасные (“проволочные”) модели»)).
Фильм «Кристаллографическая структура димера инсулина» (эпизод, показывающий общий вид белкового остова). Фильм снят в 1971 году в лаборатории Компьютерных систем университета Вашингтона.
«Электронные “коробки” Ричардса»
Развитие компьютерных систем визуализации биомолекул стало ускоряться. В 1965 году Кэрролл Джонсон (Carroll Johnson) представил программу ORTEP, способную создавать стереоизображения молекул для печати на плоттере. В середине 1970-х была получена первая кристаллографическая структура белка без построения физической модели, решённая и визуализованная целиком на компьютере [18]. При этом использовалась программа GRIP, положившая начало «электронным “коробкам” Ричардса» — классу компьютерных программ, позволяющих «вписывать» компьютерную модель структуры молекулы в экспериментально полученные карты электронной плотности без изнуряющего ручного измерения углов и расстояний, необходимого при «классическом» подходе, основанном ещё Кендрю (см. главу «Каркасные (“проволочные”) модели»). Подробнее об эволюции кристаллографических программ можно прочитать в книге Коннолли (Michael Connolly) «Молекулярные поверхности» [19].
«Новые» алгоритмы молекулярной графики
Исследования в области фолдинга белка и попытки анализа распределения гидрофобных свойств на поверхности (в контакте с растворителем) и «внутри» молекул привели к формулировке понятия «поверхности молекулы, доступной растворителю». Согласно определению, данному Ли и Ричардсом (B.K. Lee & Fred Richards, [20]), этот тип поверхности задаётся геометрическим местом центров сфер, «обкатывающих» ван-дер-ваальсову поверхность молекулы и имеющих радиус, соответствующий «эффективному» радиусу молекулы растворителя (для воды чаще всего используется значение 1,4 Å). Эта концепция, впоследствии модифицированная Коннолли, оказалась очень успешной и до сих пор чрезвычайно активно используется для визуализации поверхности биомакромолекул [[19], глава 5 — «Поверхность, доступная растворителю»].
Немного позже был разработан компьютерный алгоритм построения закрашенных поверхностей с учётом теней [21]. Это произвело целую революцию в компьютерной графике, поскольку позволяло показывать молекулы с совершенно новым уровнем наглядности, хоть в то время эта возможность была доступна лишь на самых мощных машинах.
«Новый облик» биомакромолекул стал использоваться повсеместно: в 1980 году Подразделение Компьютерных Технологий НИЗ США выпустило набор цветных учебных стереоскопических слайдов («Teaching Aids for Macromolecular Structure», TAMS) с изображением структур белков и простое устройство для их просмотра (рис. 11). В этот обучающий набор вошло 116 слайдов по темам: пептидная связь, α-спираль, β-структура, третичная и четвертичная структуры белка, кофакторы, активные сайты и др. Слайды были пересняты с экрана передового на тот момент компьютерного устройства, оперировавшего всего одним байтом информации на пиксел изображения, но даже эти 256-цветные картинки в то время выглядели очень впечатляюще.
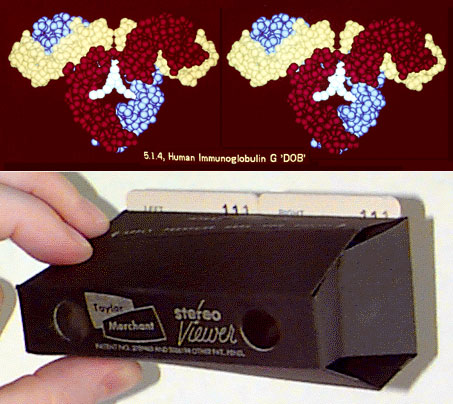
Рисунок 11. Стереослайды для образовательных целей (TAMS). Сверху: Иммуноглобулин “DOB” человека (только Cα-атомы). Лёгкая цепь показана жёлтым цветом, тяжёлые — красным и голубым. Молекула углевода окрашена в бирюзовый цвет. Снизу: Устройство для просмотра слайдов на просвет (“Tailor Merchant 101”).
Компьютеры «Эванс и Сазерленд»
В 1980-х наиболее популярной компьютерной системой среди кристаллографов была платформа, разработанная фирмой «Эванс и Сазерленд» (“Evans & Sutherland”), выполняющей кроме всего прочего многие заказы министерства обороны США (в том числе и космической тематики). Эти компьютеры, стоившие в 1985 году $250 000, умели отображать карты электронной плотности и позволяли производить ручное «вписывание» молекулярных моделей в карту. Цветной дисплей компьютера, показывавший лишь скелетное представление молекул, позволял производить вращение молекул в реальном времени. Эта система основывалась на сопроцессоре векторной графики, содержавшем несколько аппаратных блоков по работе с матрицами (перемножение, сложение векторов, расчет детерминантов и пр.). Компьютеры E&S использовали кристаллографическую программу FRODO, позже развившуюся в TURBO-FRODO и давшую начало другой популярной в 90-х годах кристаллографической программе — “O”. Один из компьютеров, выпускавшихся E&S (хотя не из числа тех, что использовались кристаллографами), показан на рис. 12.

Рисунок 12. Фотография одного из ранних компьютеров «Эванс и Сазерленд». Иван Сазерленд (Ivan Sutherland, слева) и Дэйв Эванс (Dave Evans, справа) на фоне компьютера LDS-1 (Line Drawing System—1).
Первые программы молекулярной визуализации для персональных компьютеров
Одними из пионеров «народной» молекулярной компьютерной графики являются Дэвид и Джейн Ричардсон, которые в 80-х занимались разработкой соответствующих программ в университете Дюка. В 1992-м вышла программа Kinemage (kinetic image) за авторством Ричардсонов, разработанная для компьютеров Макинтош, и получила чрезвычайно широкое распространение (поскольку эти компьютеры были несравненно более доступны «рядовым» учёным и студентам, нежели баснословно дорогие E&S). Программа, описанная в первом номере Protein Science за 1992 год [22], поставлялась на дискетке, вложенной в обложку журнала.
Вскоре после этого появилась программа RASMOL (Raster Molecules), позволявшая привнести истинную интерактивность в изучение произвольной молекулярной структуры. Роджер Сэйл (Roger Sayle), автор программы, ещё будучи студентом, написал невероятно быстрый алгоритм трассировки лучей (использующийся для построения «объёмных» молекулярных рисунков), который лёг в основу RASMOL’а. С 1990-го года Роджер под руководством кристаллографа Эндрю Кулсона (Andrew Coulson) развивал свой алгоритм, пока не адаптировал его к однопроцессорным «персоналкам» под управлением UNIX, а позднее — и Windows, и Macintosh (ранние версии его алгоритма были заточены под специализированные многопроцессорные компьютеры). В 1993 году вышла программа RASMOL (первые три буквы её названия совпадают с инициалами автора — R.A.S.), которая была сделана бесплатной вскоре после того, как Роджер защитил диссертацию, — как в виде дистрибутива, так и в виде исходных кодов. RASMOL получил очень широкое распространение, а его «исходники» были использованы в массе других программ молекулярной графики.
Заключение
На этом я намеренно заканчиваю обзор средств визуализации структуры биомакромолекул, поскольку, начиная с середины 1990-х годов, средства компьютерной молекулярной графики начали развиваться столь бурно, что описать все их направления попросту невозможно. Интересующиеся отсылаются к сайту «Каталог ресурсов молекулярной визуализации», одному из самых полных ресурсов по этой теме.
Последнее, что хотелось бы отметить, — чрезвычайная простота визуализации молекул, достигнутая в наше время (не нужно ничего, кроме самого простого компьютера и интернета), стала скорее затмевать механизмы работы молекул для рядовых пользователей программ визуализации, нежели прояснять их. Если раньше модель структуры молекулы становилась вершиной работы мысли учёного, то теперь, благодаря успехам структурной геномики и развитию компьютерных технологий, вывести на экран и покрутить молекулу белка не представляет ни малейшей сложности.
Так что, как говорил один преподаватель в Университете, «любование красивыми картинками не должно заменять понимания сути вещей».
При подготовке использовались материалы веб-сайта «История молекулярной визуализации» и книги Майкла Коннолли (Michael Connolly) «Молекулярные поверхности» [19].
Литература
- Зоркий П.М. (2001). Лайнус Полинг — величайший химик XX столетия. Химфак МГУ;
- Классический «белковый» номер журнала Proceedings of the National Academy of Sciences of U.S.A., содержащий семь статей за авторством Полинга и Кори, идущих подряд. (1951). PNAS. 5;
- Уотсон Дж.Д. Двойная спираль. Воспоминания об открытии структуры ДНК. М.: Мир, 1969;
- Нобелевские лауреаты. Лайнус Полинг. Электронная библиотека «Наука и Техника»;
- Нобелевские лауреаты. Джеймс Уотсон. Электронная библиотека «Наука и Техника»;
- Walter L. Koltun. (1965). Precision space-filling atomic models. Biopolymers. 3, 665-679;
- J. C. KENDREW, G. BODO, H. M. DINTZIS, R. G. PARRISH, H. WYCKOFF, D. C. PHILLIPS. (1958). A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature. 181, 662-666;
- M. F. PERUTZ, M. G. ROSSMANN, ANN F. CULLIS, HILARY MUIRHEAD, GEORG WILL, A. C. T. NORTH. (1960). Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis. Nature. 185, 416-422;
- HILARY MUIRHEAD, M. F. PERUTZ. (1963). Structure Of Hæemoglobin: A Three-Dimensional Fourier Synthesis of Reduced Human Haemoglobin at 5.5 Å Resolution. Nature. 199, 633-638;
- Нобелевские лауреаты. Макс Перутц. Электронная библиотека «Наука и Техника»;
- Wyckoff H.W., Hardman K.D., Allewell N.M., Inagami T., Johnson L.N., Richards F.M. (1967). The structure of ribonuclease-S at 3.5 Å resolution. J. Biol Chem. 242, 3984–3948;
- Frederic M. Richards. (1968). The matching of physical models to three-dimensional electron-density maps: A simple optical device. Journal of Molecular Biology. 37, 225-230;
- Jane S. Richardson, David C. Richardson, Kenneth A. Thomas, Enid W. Silverton, David R. Davies. (1976). Similarity of three-dimensional structure between the immunoglobulin domain and the copper, zinc superoxide dismutase subunit. Journal of Molecular Biology. 102, 221-235;
- S P Heathman, J D Nicholas, P A Timmins, J L Finney. (1976). A simple jig for building complex molecular models. J. Phys. E: Sci. Instrum.. 9, 143-145;
- Levinthal C. (1966). Molecular model-building by computer. Scientific American. 214, 42–52;
- Barry Honig. (1991). In memoriam: Cyrus levinthal. Proteins. 11, 239-241;
- R. Zwanzig, A. Szabo, B. Bagchi. (1992). Levinthal's paradox.. Proceedings of the National Academy of Sciences. 89, 20-22;
- Karl M. Beem, David C. Richardson, K. V. Rajagopalan. (1977). Metal sites of copper-zinc superoxide dismutase. Biochemistry. 16, 1930-1936;
- Connolly M. (1996). Molecular surfaces: a review. Network Science. 2;
- B. Lee, F.M. Richards. (1971). The interpretation of protein structures: Estimation of static accessibility. Journal of Molecular Biology. 55, 379-IN4;
- Thomas K. Porter. (1978). Spherical shading. Proceedings of the 5th annual conference on Computer graphics and interactive techniques - SIGGRAPH '78;
- David C. Richardson, Jane S. Richardson. (2008). The kinemage: A tool for scientific communication. Protein Science. 1, 3-9;
- Чугунов А.О. (2007). Изваяние невидимого (Молекулярная скульптура). «Компьютерра». 712, 24–26 (часть 1) и 713, 26–28 (часть 2).



Комментарии
0Чтобы оставить комментарий, необходимо
войти