Отряд самоубийц в медицине
12 сентября 2017
Отряд самоубийц в медицине
- 3721
- 0
- 7
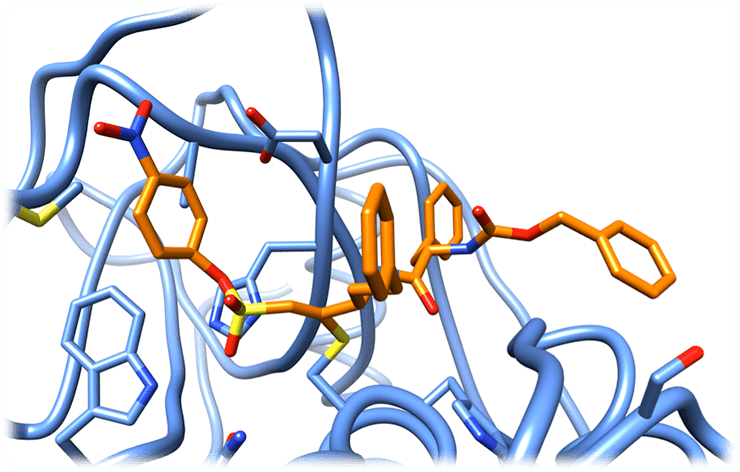
Необратимый ингибитор (оранжевый), подобно диверсанту, подрывает работу фермента (синий). Это потенциальное лекарство против трипаносомы крузи — главной причины болезни сердца в Латинской Америке.
PDB-структура 1F2B
-
Авторы
-
Редакторы
Статья на конкурс «био/мол/текст»: Многие лекарства конкурируют с природными молекулами за связывание со своей мишенью. Большинство таких препаратов связывается с белками за счет слабых взаимодействий, но некоторые способны образовывать прочные связи, «выключая» свою мишень до конца ее «жизни», пусть и ценой собственной. Такие лекарства относятся к классу необратимых ковалентных ингибиторов, получивших образное название суицидных ингибиторов (англ. suicide inhibitors). О них и пойдет речь в нашей статье. Как работают и насколько опасны одни из самых эффективных лекарств? Чья болезнь помогла открыть аспирин? Что общего между никотином и грейпфрутовым соком? Ответы на эти и многие другие вопросы вы найдете далее.

Конкурс «био/мол/текст»-2017
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2017.

Генеральный спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.

Спонсором приза зрительских симпатий и партнером номинации «Биомедицина сегодня и завтра» выступила фирма «Инвитро».

«Книжный» спонсор конкурса — «Альпина нон-фикшн»
Из чего же, из чего же сделаны эти лекарства?
Сегодня сложно представить себе человека, который ни разу в жизни не принял ни одной таблетки. Каждый год прилавки аптек пополняются все новыми и новыми лекарствами. Независимо от цвета упаковки препарата и его лекарственной формы (будь то таблетка, мазь, капсула, раствор для инъекций и т.д.) внутри всегда содержится главный компонент лекарства — действующее вещество. Именно оно и обеспечивает терапевтический эффект.
Большинство действующих веществ является ингибиторами, то есть они блокируют функцию того или иного белка (чаще фермента, но иногда и рецептора) в нашем или чужеродном организме. Многие из них связываются со своей мишенью обратимо, то есть после встраивания в мишень молекула может выйти обратно, так что их возможности сильно ограничены. Но необратимые ковалентные ингибиторы «намертво» связываются со своей мишенью. В результате фермент больше не способен функционировать до тех пор, пока клетка не синтезирует его новые копии.
Низкомолекулярные «взломщики»
Необратимые ковалентные ингибиторы — это относительно малоактивные молекулы, которые трансформируются ферментом-мишенью в высокоактивную форму, способную необратимо ингибировать (то есть «выключить») фермент [1]. Представим себе идеальную ситуацию: мы разработали ингибитор, который не обладает совершенно никакой активностью вне нашей мишени, но, попав в последнюю, узнает ее и выключает, обрекая на смерть. Но как же заставить ингибитор работать только в нужном нам белке? Ответ лежит на поверхности: использовать собственный механизм действия молекулярной мишени против самой себя. Фермент должен сперва принять ингибитор за привычную природную молекулу. Затем начать реакцию, за счет которой лекарство необратимо свяжется с мишенью [2]. То есть, приходя в качестве безобидного субстрата, этот «диверсант» намертво хватает ничего не подозревающий фермент, лишая его способности к функционированию и обрекая на верную гибель [3]! Эти вещества как бы «взламывают» нормальные ферментативные реакции для выключения белка. Правильно спроектированный необратимый ингибитор специфичен к определенному ферменту и не опасен для других молекул. Поэтому лекарства, основанные на этом подходе, могут иметь важное преимущество в виде малого количества нежелательных эффектов [1].
«В чем же секрет столь сильного связывания этих ваших необратимых ингибиторов?» — спросите вы. В ковалентной связи. Существует несколько механизмов связывания лекарств с их молекулярными мишенями. Обычно связывание происходит при помощи слабых взаимодействий [4]. Ковалентная же связь является сильнейшим из возможных взаимодействий и превосходит любые другие в десятки, а то и сотни раз (табл.1) [5]!
| Тип связи | Энергия, ккал/моль |
|---|---|
| Ковалентная | 50–150 |
| Электростатическая | 5–10 |
| Водородная | 2–5 |
| Гидрофобная | 0,5–1 |
Механизм образования ковалентной связи строится на том, что пара электронов одного из атомов молекулярной мишени (нуклеофила) распределяется между ним и одним из атомов ингибитора (электрофилом) (рис. 1). Из-за высокой прочности связи ингибитор оказывается необратимо присоединен к ферменту и приводит к разрушению последнего. Восстановление функции фермента наступает только после синтеза его новых копий [5].

Рисунок 1. Общий механизм действия необратимых ингибиторов.
Как же выглядят эти электрофильные группы, которые используются для создания ковалентных необратимых ингибиторов? Найти ответ вы сможете, посмотрев на рисунок 2. Наличие одного из этих фрагментов в структуре лекарства может говорить о том, что оно действует по уже известному вам механизму [6].

Рисунок 2. Примеры электрофильных групп, встречающихся в необратимых ингибиторах.
Несмотря на то, что связывание необратимых ингибиторов приводит к гибели фермента, не следует думать, что одна доза такого лекарства может полностью уничтожить фермент в организме. Наличие и работа генов позволит создать его новые копии, пусть на это и понадобится от нескольких часов до пары дней [2].
Как и все в мире, ковалентные необратимые ингибиторы имеют свои преимущества и недостатки, которые мы представили в формате таблицы 2 [6].
| Преимущества | Недостатки |
|---|---|
| Высокие эффективность и избирательность позволяют использовать меньшие дозы и уменьшить риск побочных эффектов | Высокая активность некоторых представителей может приводить к некрозу печени, мутациям и даже раку! |
| Связывание с важными остатками фермента предотвращает развитие устойчивости микробов, что важно при лечении инфекций | Потенциальная иммуногенность, так как полученный продукт реакции может вызывать аллергический ответ |
| Длительное действие, так как активность фермента вновь появляется только при синтезе его новых копий |
Тернистые пути разработки эффективных лекарств
Если вы думаете, что разработка необратимых ковалентных ингибиторов — легкое дело, то вы глубоко заблуждаетесь. Даже если закрыть глаза на то, что разработка любого лекарства является крайне трудоемким и времязатратным процессом [7], задача не сильно упростится — ведь необратимые ингибиторы требуют особого подхода к своей персоне. Сегодня не так много одобренных препаратов действуют именно таким образом. А механизм действия многих из них открыли только спустя годы после разработки! Ученые на этом пути сталкиваются с большим количеством порой неразрешимых проблем.
Одна из них — участие одного фермента сразу в нескольких метаболических каскадах. Обсуждаемые лекарства необратимо связываются с ферментом-мишенью, уничтожая его на то время, которое требуется на синтез новых копий фермента. Этот процесс может занимать до нескольких суток! В том случае, если фермент-мишень выполнял несколько функций, это чревато серьезными побочными эффектами. Чтобы обойти проблему, необратимо связывающиеся ингибиторы чаще всего применяют для блокирования одного из ферментов потенциально опасных для человека вирусов, бактерий и т. д. [3].
Вторая проблема заключается в том, что при низкой избирательности необратимые ингибиторы могут связываться с нежелательными мишенями, похожими на выбранную нами для ингибирования. Это тоже может приводить к серьезным побочным эффектам. Поэтому важная задача при оптимизации структуры ингибиторов — повышение избирательности к своей мишени [3].
Опасения в отношении побочных эффектов необратимых ингибиторов не лишены оснований. Бум гонений на эту группу препаратов вызвали исследования 1970-х годов. Тогда выяснили, что два из наиболее широко известных представителей этой группы — парацетамол и фуросемид — обладают высокой токсичностью в связи с тем, что, метаболизируясь в печени, они образовывают активные производные, формирующие ковалентные связи с ее белками [2], [8]. Как выяснилось позже, эти единичные случаи не имеют отношения ко многим препаратам из группы необратимых ингибиторов, а парацетамол и фуросемид до сих пор активно используются в клинической практике.
Другим распространенным примером нежелательного необратимого ингибирования является цитохром P450. Цитохромы — группа ферментов в нашем организме, главной задачей которых является обезвреживание потенциально опасных веществ. Первыми необратимыми ингибиторами цитохрома стали алкены и алкины. В результате ингибирования этого фермента процессы детоксикации становятся невозможными. Неполный список препаратов, способных вступать в подобную реакцию, включает 17α-алкенил-стероиды, левомицетин, циклофосфамид, спиронолактон, фурокумарины, никотин, изониазид и барбитураты. Звучит опасно, не правда ли? Не многим опаснее, чем пить грейпфрутовый сок! Его компоненты способны необратимо связываться с цитохромом кишечника и лишать этот фермент активности на 24 часа! К чему это может привести? К тому, что принимаемые в это время лекарства будут более эффективно поступать в наш организм, а рост их концентрации повышает риск развития нежелательных эффектов [5]. Именно поэтому врачи рекомендуют запивать таблетки только водой.
Но всегда ли можно считать необратимые ингибиторы безопасными лекарствами? Конечно же, нет. Поиск необратимых ингибиторов, и правда, не всегда оборачивается успехом для исследователей. В частности, полным провалом завершились исследования мышьяк-содержащих органических соединений для лечения трипаносомоза [9]. Эти соединения необратимо ингибировали жизненно необходимые ферменты трипаносомы за счет образования связей между атомом мышьяка в составе исследуемых веществ и серосодержащими остатками фермента. Но они оказались токсичными не только для трипаносомы, но и для организма человека [5].
К счастью, несмотря на все перипетии судьбы, ученые вновь сфокусировали свои пристальные взгляды на необратимых ковалентных ингибиторах. Лучшее понимание реально существующих рисков и знание механизма химических преобразований открыли множество возможностей для разработки более эффективных лекарств и привели к резкому росту встречаемости необратимых ингибиторов в различных исследованиях [10].
«Ветераны» борьбы за здоровье человечества
Многие из широко применяемых успешных препаратов — необратимые ковалентные ингибиторы [6]. Например, в 2009 году три необратимо связывающихся ингибитора попали в топ-10 наиболее продаваемых лекарств [2]. В 2011 году из 39 ковалентных лекарств, одобренных в США, 33% являлись противоинфекционными, 20% нашли применение в лечении рака, 15% применялись при болезнях пищеварительного тракта, 10% — при нарушении работы центральной нервной системы, 5% использовались при лечении сердечно-сосудистых заболеваний и один препарат обладал противовоспалительными свойствами (рис. 3) [6], [11].

Рисунок 3. Области применения одобренных в США необратимо связывающихся ингибиторов (данные за 2011 год).

Рисунок 4. Структура салицилата натрия (а) и аспирина (б).
Давайте же поговорим о нескольких наиболее успешных примерах необратимых ковалентных ингибиторов.
Натрия салицилат впервые применили в качестве противовоспалительного лекарства в 1875 году (рис. 4а). Препарат обладал ужасным вкусом и вызывал язвы ротовой полости и желудка. В конце XIX века отец одного из химиков компании Bayer Company, страдающий от тяжелого ревматоидного артрита, упросил своего сына, Феликса Хоффмана, заняться поиском менее опасного аналога салицилата натрия. Феликс синтезировал различные производные и обнаружил, что ацетилсалициловая кислота обладает лучшими свойствами. Так был обнаружен аспирин (рис. 4б). В 1899 году Bayer выпустила аспирин как противовоспалительное, обезболивающее и жаропонижающее средство [2]. Как и в случае многих других препаратов, механизм его действия открыли только спустя 70 лет после коммерциализации препарата [6], за что в 1982 году была вручена Нобелевская премия [2], [12]. Аспирин оказался необратимым ингибитором ферментов, участвующих в реакции воспаления — циклооксигеназ [6].

Рисунок 5. Структура пенициллина.
Другие примеры необратимых ковалентных ингибиторов — пенициллины [6]. Их родоначальника, пенициллин (рис. 5), открыл в 1928 году Александр Флеминг [13]. Ученым понадобилось еще 15 лет, чтобы достаточно изучить и начать использовать этот препарат в качестве антибактериального лекарственного средства, что спасло миллионы жизней во время Второй мировой войны [12]. Пенициллин нарушает синтез клеточной стенки бактерий. Он связывается с транспептидазой — ферментом, обеспечивающим первый этап сшивки пептидогликана, который является основным компонентом бактериальной клеточной стенки. Когда пенициллин связывается с транспептидазой, синтез бактериальной клеточной стенки блокируется, и многие бактерии погибают от разрыва клеточной мембраны под воздействием осмотического давления [1].
Однако все оказалось не так радужно — у бактерий обнаружились механизмы устойчивости [14]. В частности, резистентность к пенициллинам обеспечивается за счет наличия β-лактамаз — ферментов, расщепляющих пенициллины. Ученые не остановились на достигнутом и разработали ингибиторы этого фермента — сульбактам, тазобактам и клавулановую кислоту. Подобно пенициллинам, в результате нуклеофильной атаки эти соединения претерпевают открытие β-лактамного кольца. Отличие в том, что ингибиторы β-лактамаз подвергаются раскрытию и второго цикла, содержащегося в их структуре. При этом появляется возможность образования дополнительных связей, в том числе и ковалентных (рис. 6) [6], [15].

Рисунок 6. Механизм действия ингибиторов β-лактамаз

Рисунок 7. Структура ацикловира.
Ацикловир — один из самых эффективных препаратов с противовирусной активностью (рис. 7). Он был открыт в 1974 году крупной ныне фармацевтической компанией — GlaxoSmithKline (тогда она называлась Burroughs Wellcome) — в ходе широкомасштабного поиска противовирусного средства, начавшегося еще в 1960-е годы. А в 1988 году за изучение механизмов действия ацикловира и других препаратов вручили Нобелевскую премию [16].
В клинической практике ацикловир применяют для лечения вируса герпеса. Важно, что здоровые клетки человека не подвергаются действию препарата. Ведь ацикловир попадает в клетку благодаря одному из ферментов вируса — тимидинкиназе! Способность ацикловира связываться с этим ферментом в 200 раз выше, чем с любым ферментом нашего организма. Оказавшись в зараженных клетках, ацикловир преобразуется в ацикловир трифосфат, который связывается с вирусной ДНК и в таком виде необратимо ингибирует вирусные ДНК-полимеразы, необходимые для размножения вируса [17].
Другим широко известным примером необратимых ингибиторов является омепразол, одобренный к применению в 1980-х. Его применяют при заболеваниях пищеварительной системы, сопровождающихся усиленным выделением желудочной кислоты, таких как изжога или язва желудка. В нейтральной среде омепразол не оказывает действия на наш организм, но при ее закислении желудочным соком необратимо связывается с протонной помпой, ответственной за образование кислоты в желудке. Это происходит потому, что благодаря кислой среде омепразол претерпевает ряд внутримолекулярных перестроек и становится способным принять на себя нуклеофильную атаку каталитического цистеина протонной помпы, тем самым образуя прочную ковалентную связь и выключая этот белок (рис. 8) [6], [18], [19].

Рисунок 8. Механизм действия омепразола.
Африканская сонная болезнь, или африканский трипаносомоз, вызывается простейшими рода трипаносома. До конца XX века эта болезнь была практически неизлечима. Вакцины оказались неэффективны против трипаносом из-за того, что последние имеют механизмы обхода иммунной системы человека. Поэтому, основываясь на знаниях о жизненно необходимых ферментах этих простейших, ученые прибегли к разработке механизм-опосредованно связывающихся ингибиторов. Ахиллесовой пятой в метаболизме трипаносом оказался путь биосинтеза полиаминов, вовлеченных в процессы упаковки ДНК и в больших количествах необходимых при делении клеток. Первый этап их синтеза обеспечивает фермент орнитиндекарбоксилаза при участии вспомогательной молекулы — витамина В6. В клетках млекопитающих фермент синтезируется быстро, а у трипаносом этот процесс занимает гораздо больше времени. Именно поэтому необратимые ингибиторы орнитиндекарбоксилазы мало влияют на человеческие клетки, но крайне губительно действуют на паразита [1]. На базе этого механизма был разработан дифторметилорнитин (ДФМО). ДФМО достаточно инертен в растворе, но в результате реакции с витамином В6 приобретает способность связываться с орнитиндекарбоксилазой, которая быстро инактивируется (рис. 9). Эта реакция необратима. Таким образом ДФМО использует собственные реакции фермента для его уничтожения. Препарат показал высокую эффективность против африканской сонной болезни в клинических исследованиях и используется в настоящее время для ее лечения [1].

Рисунок 9. Комплекс производного ДФМО (голубой) с витамином В6 (зеленый) и ферментом орнитиндекарбоксилазой (серый).
PDB-структура 2TOD.
А недавно ученые обнаружили, что ДФМО также может использоваться в качестве препарата против рака! Механизм действия ДФМО по отношению к раковым клеткам хорошо изучен. Так же, как и в случае лечения трипаносомоза, этот агент ингибирует действие орнитиндекарбоксилазы. Ведь, что интересно, в опухолевых клетках количество этого белка значительно выше, так как они интенсивно делятся! При инактивации орнитиндекарбоксилазы в раковых клетках падает уровень полиаминов, что нарушает правильную упаковку ДНК, в результате чего клетки перестают делиться. Этот эффект препарата называется цитостатическим. Однако если препарат применять долго, то происходит не замедление деления, а гибель раковых клеток! ДФМО сам по себе не обеспечивает достаточный противораковый эффект, но в комбинации с другими химиотерапевтическими препаратами этот компонент показал увеличение выживаемости пациентов и ускорение темпов выздоровления. Поэтому ДФМО имеет потенциал применения в качестве лекарства, предотвращающего возникновение онкологии, либо снижающего вероятность рецидива рака после удаления опухолевых клеток [20].
Есть ли будущее у необратимых ковалентных ингибиторов?
Долгие годы необратимые ингибиторы были окружены аурой скептицизма, возникшей из-за токсичности некоторых представителей этой группы, обнаруженной в 1970-е годы. Но эффективность и избирательность многих препаратов этой серии доказали, что страхи в их отношении не были оправданными. Конечно, они обладают потенциальной опасностью из-за возможности неспецифических взаимодействий и аллергических реакций, но клиническая практика показывает их незаменимость в лечении целого ряда заболеваний. Сейчас число препаратов этой группы неуклонно растет [6].
Сложность в том, что исследователь должен преследовать сразу несколько целей: повысить избирательность лекарства к ферменту-мишени и тщательно продумать механизм его действия. Но такие свойства необратимых ковалентных ингибиторов, как высокие избирательность и эффективность, высокая продолжительность действия, сниженный риск развития устойчивости к препарату, делают эту группу лекарств незаменимым инструментом в арсенале врачей и разработчиков лекарств [6].
Литература
- Nelson D.L. and Cox M.M. Lehninger principles of biochemistry (6 Edition). Freeman W.H., 2012. — 1340 p.;
- Silverman R.B. and Holladay M.W. The organic chemistry of drug design and drug action (3rd Edition). San Diego, CA: Academic Press, 2014. — 536 p.;
- Young D.C. Computational drug design: a guide for computational and medicinal chemists (1 Edition). Wiley-Interscience, 2009. — 344 p.;
- Роль слабых взаимодействий в биополимерах;
- Lemke T.L. and Williams D.A. Foye’s principles of medicinal chemistry. Lippincott Williams & Wilkins, 2012. — 1520 p.;
- Concepción González-Bello. (2016). Designing Irreversible Inhibitors-Worth the Effort?. ChemMedChem. 11, 22-30;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Smith D.A. Metabolism, pharmacokinetics and toxicity of functional groups impact of chemical building blocks on ADMET. Cambridge: Royal Society of Chemistry, 2010. — 544 p.;
- Компьютеры против сонной болезни;
- Renato A. Bauer. (2015). Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today. 20, 1061-1073;
- Juswinder Singh, Russell C. Petter, Thomas A. Baillie, Adrian Whitty. (2011). The resurgence of covalent drugs. Nat Rev Drug Discov. 10, 307-317;
- Кузница нобелевских кадров;
- Победитель бактерий;
- Противостояние с резистентными бактериями: наши поражения, победы и планы на будущее;
- S. M. Drawz, R. A. Bonomo. (2010). Three Decades of -Lactamase Inhibitors. Clinical Microbiology Reviews. 23, 160-201;
- King D.H. (1988). History, pharmacokinetics, and pharmacology of acyclovir. J. Am. Acad. Dermatol. 18, 176–179;
- Beale J.M. and Block J. Wilson and Gisvold’s textbook of organic medicinal and pharmaceutical chemistry (12 Edition). Wolters Kluwer, 2010. — 1008 p.;
- Fellenius E., Elander B., Wallmark B., Helander H.F., Berglindh T. (1982). Inhibition of acid secretion in isolated gastric glands by substituted benzimidazoles. Am. J. Physiol. 243, 505–510;
- Lars Olbe, Enar Carlsson, Per Lindberg. (2003). A proton-pump inhibitor expedition: the case histories of omeprazole and esomeprazole. Nat Rev Drug Discov. 2, 132-139;
- George A Alexiou, Georgios D Lianos, Vassileios Ragos, Vasiliki Galani, Athanassios P Kyritsis. (2017). Difluoromethylornithine in cancer: new advances. Future Oncology. 13, 809-819.



Комментарии
0Чтобы оставить комментарий, необходимо
войти