Лучше один раз увидеть, или Микроскопия сверхвысокого разрешения
01 ноября 2012
Лучше один раз увидеть, или Микроскопия сверхвысокого разрешения
- 12370
- 0
- 9

Виментин и клатрин, меченные флуоресцентными антителами. Изображение слева получено с помощью традиционного конфокального микроскопа, справа — с применением STED-технологии. С помощью методов сверхвысокого разрешения можно различить структуры до 10 нм (так, справа различимы фибриллы отдельных промежуточных филаментов).
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: Благодаря зрению мы получаем 90% информации об окружающем нас мире. Именно поэтому микроскопия играет огромную роль в различных направлениях современной биологии. Долгое время дифракционный барьер не позволял изучать структуры менее 200 нм, но сейчас удалось найти сразу несколько решений данной проблемы.

Конкурс «био/мол/текст»-2012
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2012 в номинации «Лучший обзор».
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Разрешение стандартного оптического микроскопа
Уже с конца XVI века ученые начали применять увеличительные стекла и конструировать первые микроскопы (Ханс Янсен, Галилео Галилей, Корнелиус Дреббель, Кристиан Гюйгенс, Роберт Гук и Антони ван Левенгук), чтобы как можно более подробно изучить тончайшую структуру жизни. Постоянное совершенствование оптических микроскопов привело к тому, что на сегодня достигнуто увеличение более чем в 2000 раз [1]. Можно было бы и еще больше, но дальнейшее увеличение просто не имеет смысла, поскольку оно не поможет различить более мелкие детали препарата.
Поясним, что не следует путать увеличение микроскопа и его разрешающую способность. Так, увеличение определяет, во сколько раз изображение, построенное оптической системой микроскопа, больше самого объекта, а разрешающая способность определяет то минимальное расстояние, на котором независимые источники света будут различимы. Разрешающая способность микроскопа, как было установлено в 1873 году Эрнстом Аббе, характеризуется неким предельным значением, обусловленным волновой природой света. Свет от точечного источника (размеры которого значительно меньше длины световой волны), проходя через оптическую систему, формирует не точку, а светлый кружок с темными и светлыми кольцами (дифракция Фраунгофера или дифракция в параллельных лучах). Функция, характеризующая трехмерное распределение интенсивности света в таком изображении, называется функцией рассеяния точки. На центральный кружок (диск Эйри) приходится 85% интенсивности света, и именно из таких кружков складывается изображение в оптической микроскопии (рис. 1а).
Если два точечных источника света расположены ближе некоторого критического значения, то их изображения (диски Эйри) будут перекрываться, и их невозможно идентифицировать как отдельные светящиеся точки (рис. 1б) [2], [3]. Это минимальное расстояние и есть дифракционный предел, который рассчитывается по формуле d = 0,61λ/NA, где NA = nsinα — нумерическая апертура объектива, n — коэффициент преломления среды, α — угол между оптической осью объектива и наиболее отклоняющимся лучом, попадающим в объектив (апертурный угол), λ — длина световой волны (рис. 1в). В связи с тем, что объект освещается только с одной стороны, разрешение вдоль оптической оси еще меньше: d = 2λn/(NA)2 [4], [5]. В микроскопии в видимом свете с масляной иммерсией и NA = 1,4 можно достичь максимального разрешения около 200 нм в латеральной плоскости и 500 нм — в аксиальной [6].

Рисунок 1. Дифракционный предел. а — Изображение точечного источника света, сформированное оптической системой микроскопа. На центральный максимум приходится приблизительно 85% интенсивности от всех частей изображения. б — Точечные источники света, расположенные на различном расстоянии друг от друга. Расстояние между источниками 1 и 2 значительно больше d (дифракционного предела). Расстояние между 3 и 4 равно d/2, а между 5 и 6 значительно меньше d/2. в — Изображения двух точечных источников света, полученные объективами с одинаковыми числовыми апертурами (то есть, с одинаковым разрешением), но дающие различное увеличение. Расстояние между источниками составляет d/2. Очевидно, что дополнительное увеличение не позволяет получить более четкую картину.
журнал «Квант»
Технические хитрости

Рисунок 2. Основные параметры, определяющие разрешение объектива: n — коэффициент преломления иммерсионной жидкости и α — апертурный угол объектива.
Очевидно, что такого разрешения недостаточно для детального изучения структуры и процессов, происходящих на субклеточном уровне. Так, например, размеры рибосом, ядерных пор, АТФ-синтаз, клеточных филаментов, микротрубочек и других надмолекулярных структур не превышают 150 нм. Толщина биологических мембран составляет не более 10 нм. Электронная микроскопия позволяет достигнуть необходимого разрешения, но она не пригодна для работы с живыми клетками из-за высокой разрушающей и ионизационной способности, а также предполагает напыление тонких слоев металла или углерода, что может изменить исходные свойства объекта. Атомно-силовая микроскопия «близорука» и не позволяет проникнуть вглубь объекта более чем на 10–20 нм.
Взамен атомно-силовая микроскопия, конечно, обладает другими уникальными свойствами: «Атомно-силовая микроскопия: увидеть, прикоснувшись» [15]. — Ред.
Важной особенностью живых клеток и тканей является низкий контраст внутренних структур, которые в основном прозрачны. Для их идентификации необходимо специфическое окрашивание, в том числе с применением различных флуорофоров (органических молекул, флуоресцентных белков или квантовых точек). Таким образом, флуоресцентная микроскопия сочетает в себе сразу несколько преимуществ. Во-первых, возможность прижизненного изучения объектов и наблюдения процессов в реальном времени. Во-вторых, возможность специфического мечения тканей, клеток, органелл и отдельных молекул. В-третьих, доступное на данный момент разнообразие флуоресцентных красителей и белков позволяет изучать одновременно несколько мишеней [6].
Общая концепция применения флуоресцентных методов в молекулярно-биологических исследованиях изложена в статье «Рулетка для спектроскописта» [16]. На сайте Института биоорганической химии РАН можно посмотреть видеозаписи трех докладов, объединенных общей темой «Флуорофоры: органические, неорганические и генетически кодируемые». — Ред.
Существует достаточно много технологий, основанных на различных физических феноменах, позволяющих увеличить разрешение как в латеральном, так и аксиальном направлениях. Ближнепольная сканирующая микроскопия (микроскопия без использования линз) преодолевает дифракционный предел (разрешение порядка 20–50 нм), но позволяет изучать только поверхностные свойства объекта [6]. Однако биологические объекты трехмерны, и с увеличением толщины объекта свет от разных слоев будет затруднять интерпретацию изображения конкретного оптического среза. Известно несколько методов микроскопии дальнего поля, значимость которых в первую очередь определяется заметным улучшением разрешения вдоль оси Z.
В конфокальном микроскопе применяется апертура в фокальной плоскости объектива, пропускающая свет только от объектов, находящихся в фокусе [7]. Мультифотонная микроскопия основана на возможности двухфотонного или трехфотонного возбуждения флуоресценции. Например, флуорофор, обычно поглощающий ультрафиолетовое излучение (≈350 нм), может быть возбужден двумя красными фотонами (≈700 нм), если они достигли флуорофора одновременно (поглощение будет зависеть от квадрата интенсивности возбуждающего излучения). Это значит, что необходима высокая плотность фотонов для возбуждения флуоресценции. Достаточная плотность достигается в фокусе, поэтому возбуждение флуоресценции происходит только в фокальной плоскости [8]. В I5M- и 4Pi-микроскопии применяются два объектива для возбуждения и/или регистрации флуоресценции, что позволяет освещать и регистрировать флуоресценцию с двух сторон от образца и заметно увеличить разрешение вдоль оптической оси (до 100 нм) [6].
Еще одним интересным методом является микроскопия структурированного освещения — SIM (Structured Illumination Microscopy). В плоскости, сопряженной с фокальной, располагается решетка, создающая определенный паттерн освещения. Индуцируемая флуоресценция повторяет паттерн освещения, при этом флуоресценция объектов, расположенных в фокальной плоскости, сильно меняется при перемещении этого паттерна. Флуоресценция объектов, расположенных не в фокусе, от сдвига решетки практически не зависит. Последующая компьютерная обработка позволяет отсечь флуоресценцию от остальных оптических слоев и также улучшить разрешение вдоль оси Z (рис. 3). В HR-SIM (High Resolution SIM) на образец проецируется освещение, характеризующееся высокой периодичностью. При взаимодействии с неизвестной структурой объекта, получается изображение с периодом выше, чем у двух изначально взаимодействующих образцов решетки и исследуемого объекта (эффект муара). Несколько раз изменяя положение решетки и анализируя различные изображения с муаровым эффектом, можно воссоздать исходную структуру объекта, недоступную обычной микроскопии из-за дифракционного предела [9].

Рисунок 3. Микроскопия структурированного освещения. На трех исходных изображениях (а—в) видно, что при перемещении решетки (обозначено черной линией) интенсивность флуоресценции объектов, расположенных в фокусе, заметно меняется. Флуоресценция, исходящая от других оптических слоев, практически не меняется (обозначено белой стрелкой), что позволяет избавиться от нее за счет компьютерной обработки (г).
Все перечисленные выше методы не преодолевают дифракционный предел как таковой. Сочетая сразу несколько из них, вдоль осей XYZ возможно увеличить разрешение максимум в два раза, но оно по-прежнему будет зависеть от λ и α [4].
Микроскопия сверхвысокого разрешения
Таким образом, из-за дифракции вместо точечного изображения флуорофора получается размытое пятно. Однако дифракция не препятствует более точному определению координат данного флуорофора, если в его окрестности не находятся другие источники флуоресценции. Если флуоресцентные молекулы можно обратимо переводить из флуоресцентного состояния А в темновое состояние В так, чтобы молекулы в состоянии А были окружены молекулами в состоянии В, координаты молекул в состоянии А можно определить достаточно точно. Последовательно регистрируя некоторый пул молекул в состоянии А и запоминая в каждом считывании их координаты, из этих данных можно реконструировать изображение с субдифракционным разрешением [10].
Один из способов определить точное положение флуоресцирующих молекул в некоторой точке — целенаправленно перевести флуорофоры вокруг этой точки в темновое состояние [10]. Данный подход реализован в группе методов RESOLFT (REversible Saturable OpticaL Fluorescence Transitions), объединяющей несколько похожих концепций [4]. Первая практически реализованная из них — это микроскопия STED (STimulated Emission Depletion, метод подавления спонтанного испускания), основанная на подавлении эмиссии флуорофоров, расположенных вне центра возбуждения. Когда флуорофор находится в возбужденном состоянии А и встречает фотон с энергией, соответствующей разнице энергий между возбужденным и основным состоянием В, он возвращается в основное состояние до того, как произойдет спонтанная флуоресценция (рис. 4а). Для вынужденной эмиссии флуорофоры освещаются кроме возбуждающего света STED-лазером с особым пространственным распределением интенсивностей в виде «пончика» с нулевой интенсивностью в центре. В результате флуоресцируют только молекулы, расположенные близко к области с нулевой интенсивностью STED-лазера, что сужает размер функции рассеяния точки (рис. 4б, в). Далее последовательно происходит сканирование всего образца (рис. 5) [4].
О применении STED-микроскопии для изучения субмикроскопических неоднородностей в липидных мембранах клеток и визуализации распределения некоторых мембранных белков между жидкокристаллической и «рафтовой» фазами мембраны мы писали в статье «Липидный фундамент жизни» [17]. — Ред.

Рисунок 4. STED-микроскопия. а — Процесс вынужденной и спонтанной эмиссии. Когда флуорофор поглощает фотон возбуждающего света, он переходит из основного состояния S0 в возбужденное S1. Спонтанная эмиссия происходит, когда флуорофор возвращается в основное состояние. Вынужденная эмиссия происходит, если флуорофор поглощает фотон с энергией, сравнимой с разницей между основным и возбужденным состоянием. б — Схематическое изображение STED-микроскопа: свет от возбуждающего и STED-лазера одновременно фокусируются на одном участке образца. в — Распределение интенсивностей возбуждающего лазера и STED-лазера, который подавляет спонтанную флуоресценцию вокруг нулевой точки. В результате сужается функция рассеяния точки.

Рисунок 5. Две стратегии получения изображений со сверхвысоким разрешением. При направленном считывании данных (а) каждая точка образца облучается светом с особым распределением интенсивностей, так что все молекулы вокруг данной точки оказываются в темновом состоянии. Это уменьшает размеры участка с молекулами во флуоресцентном состоянии и, следовательно, функцию рассеяния точки. Для увеличения скорости сканирования образца можно применить распределение интенсивностей в виде линий с максимумами и нулевыми значениями (правый нижний угол). Для высокого разрешения по всем направлениям необходимо несколько раз повернуть паттерн освещения. При стохастическом считывании данных (б) индивидуальные молекулы переключаются между флуоресцентным и темновым состоянием. Интенсивность возбуждающего света подобрана так, что флуоресцирующие молекулы окружены молекулами в темновом состоянии. Если флуорофор яркий, можно очень точно определить его локализацию. в — Сравнение изображений, полученных с помощью оптической микроскопии и технологий сверхвысокого разрешения: слева — меченный антителами виментин, справа — микротрубочки (зеленые) и пероксисомы (красные) в клетках млекопитающих.
Метод STED позволят получить субдифракционное разрешение, рассчитываемое по формуле
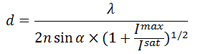
где Imax — применяемая интенсивность STED-лазера, Isat — интенсивность, которая необходима для 50% вынужденной эмиссии. Увеличение Imax до высоких значений способствует быстрому фотовыцветанию образца, поэтому для увеличения разрешения можно уменьшить Isat, которая обратно пропорциональна времени жизни флуорофора. Для этого можно изменить природу флуоресцентного состояния А и темнового В [11]. В методе STED S0 — основное, а S1 — возбужденное флуоресцентное состояние. В других методах переход из флуоресцентого состояния в темновое может представлять фотохимическую реакцию с цис-транс изомеризацией хромофора флуоресцентного белка или переход между флуоресцентным и триплетным состояниями флуорофора [10].
Принципиально другой подход основан на последовательной стохастической активации флуорофоров. Если за каждый раунд активации небольшое число флуорофоров переходит в флуоресцентное состояние А, и при этом интенсивность света подобрана таким образом, что активированные флуорофоры находятся на расстоянии примерно 200 нм, то положение каждого флуорофора можно определить с точностью до 1 нм. Далее активированные флуорофоры «выключают».

Многократное повторение циклов активации позволяет реконструировать изображении со сверхвысоким разрешением (рис. 5), где х — точность локализации (параметр, соответствующий разрешению в традиционной микроскопии), k1 и k2 определяются длиной волны возбуждаемого света, нумерической апертурой объектива и размерами пикселя, b — уровень шума на пиксель и N — число испущенных фотонов. Исходя из формулы, для данного типа микроскопии предпочтительны яркие флуорофоры с высоким коэффициентом экстинкции и квантовым выходом [11].
Идея случайной последовательной активации флуорофоров лежит в основе методов STORM (STochastic Optical Reconstruction Microscopy), PALM (PhotoActivated Localization Microscopy), и FPALM (Fluorescence PhotoActivation Localization Microscopy), разработанных тремя независимыми лабораториями [4].
При изучении 3D-структуры биологических объектов для построения изображения применяют слабые цилиндрические линзы. У таких линз фокальные плоскости в направлениях X и Y немного отличаются. Таким образом, эллиптичность и ориентация изображения флуорофора зависит от его положения по оси Z. Когда флуорофор находится в средней фокальной плоскости (примерно посередине между фокальными плоскостями для латеральных направлений X и Y), то функция рассеяния точки изодиаметрична (имеет одинаковую длину по осям X и Y). Когда флуорофор расположен выше фокальной плоскости, изображение более сфокусировано вдоль оси Y, чем оси X, поэтому оно выглядит не круглым, как в предыдущем случае, а овальным (вытянутым вдоль оси X). И наоборот, когда флуорофор находится ниже средней фокальной плоскости, функция рассеяния точки оказывается вытянутой вдоль оси Y. Анализируя форму полученных изображений, можно установить не только координаты флуорофора по осям X и Y, но и однозначно определить положение относительно оси Z (рис. 6) [13].

Рисунок 6. Принцип микроскопии 3D-STORM. Трехмерная локализация индивидуальных флуорофоров с использованием цилиндрических линз. На диаграмме справа показано, как связаны эллиптичность изображения флуорофора и его Z-координата.
Дифракционный предел преодолен!
И, наконец, необходимо сказать, что недавно физики открыли способ непосредственно преодолеть дифракционный предел. Для этого необходимо сконструировать микроскоп с линзами из метаматериала — материала с отрицательным коэффициентом преломления. Веществ с такими свойствами в природе не обнаружено, их можно получить только искусственным путем в лаборатории. Существование таких веществ было предсказано еще 40 лет назад советским физиком Виктором Веселаго, а созданы они были только в 2000-х годах. Так, отрицательный коэффициент преломления означает, что преломленный луч в среде с отрицательным коэффициентом находится с той же стороны, что и падающий (обычно падающий и преломленный лучи находятся с разных сторон от нормали, поведенной к границе раздела сред). Поэтому плоский брусок такого материала может выполнять роль суперлинзы, позволяющей различить детали по размерам меньшие полудлины волны [14].
Литература
- Википедия: «Оптический микроскоп»;
- Сердюк И., Заккаи Н., Заккаи Дж. Методы в молекулярной биофизике: структура, функция, динамика. Изд. КДУ, 2010;
- Штейн Г.И. Руководство по конфокальной микроскопии. Изд. ИНЦ РАН, 2007;
- Stefan W Hell, Marcus Dyba, Stefan Jakobs. (2004). Concepts for nanoscale resolution in fluorescence microscopy. Current Opinion in Neurobiology. 14, 599-609;
- Alexander Egner, Stefan W. Hell. (2005). Fluorescence microscopy with super-resolved optical sections. Trends in Cell Biology. 15, 207-215;
- Bo Huang, Mark Bates, Xiaowei Zhuang. (2009). Super-Resolution Fluorescence Microscopy. Annu. Rev. Biochem.. 78, 993-1016;
- Феофанов А.В. (2007). Спектральная лазерная сканирующая конфокальная микроскопия в биологических исследованиях. «Успехи биол. химии». 47, 371–410;
- D Piston. (1999). Imaging living cells and tissues by two-photon excitation microscopy. Trends in Cell Biology. 9, 66-69;
- Matthias F. Langhorst, Joerg Schaffer, Bernhard Goetze. (2009). Structure brings clarity: Structured illumination microscopy in cell biology. Biotechnol. J.. 4, 858-865;
- S. W. Hell. (2007). Far-Field Optical Nanoscopy. Science. 316, 1153-1158;
- Marta Fernández-Suárez, Alice Y. Ting. (2008). Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol. 9, 929-943;
- Stefan W Hell. (2009). Microscopy and its focal switch. Nat Methods. 6, 24-32;
- B. Huang, W. Wang, M. Bates, X. Zhuang. (2008). Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science. 319, 810-813;
- Элементы: «В поисках суперлинзы»;
- Атомно-силовая микроскопия: увидеть, прикоснувшись;
- Рулетка для спектроскописта;
- Липидный фундамент жизни.



Комментарии
0Чтобы оставить комментарий, необходимо
войти