Виртуальные тропы реальных лекарств
28 ноября 2014
Виртуальные тропы реальных лекарств
- 4127
- 0
- 11
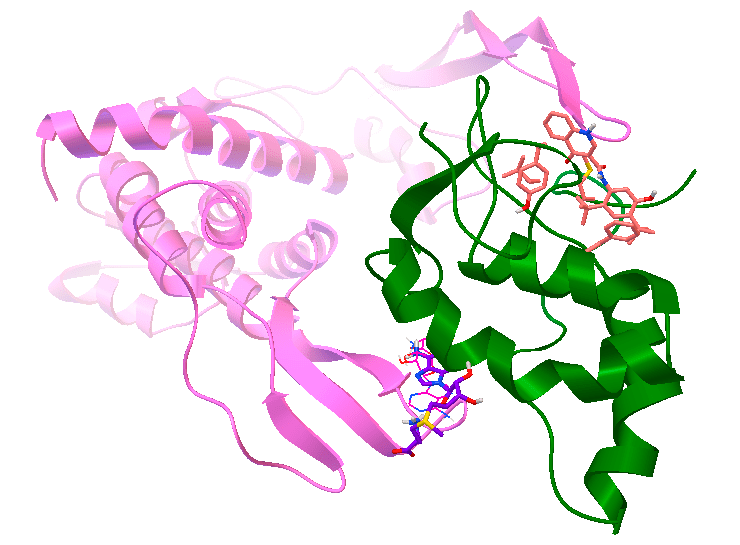
Результаты использования молекулярного докинга [2] для поиска малой молекулы (лиганда), которая бы помешала взаимодействию белков Wnt (розовый) и Frizzled (зеленый), вследствие чего могла бы быть эффективной в борьбе с раком молочной железы, в развитии которого важную роль играет взаимодействие этих белков [13]. Рисунок сделан в программе AutoDock Tools с использованием результатов работы программы Vina AutoDock.
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: В наше время компьютерные методы используются практически во всех областях науки. Создание новых лекарств не является исключением. Многие вещества, которые легко получить из природы, например, выделив экстракт из листьев какого-нибудь растения, уже давно исследованы на наличие лекарственной активности. Поэтому сейчас лекарства часто «изобретают» на компьютере, а потом уже проверяют экспериментально. В статье обсуждаются различные аспекты метода молекулярного докинга, используемые в решении этой задачи.

Конкурс «био/мол/текст»-2014
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2014 в номинации «Биоинформатика и молекулярная эволюция».

Главный спонсор конкурса — дальновидная компания «Генотек».
Конкурс поддержан ОАО «РВК».
Спонсором номинации «Биоинформатика» является Институт биоинформатики.
Спонсором приза зрительских симпатий выступила фирма Helicon.
Свой приз также вручает Фонд поддержки передовых биотехнологий.
Спонсор публикации этой статьи — Лев Макаров.
Далеко не всегда лучшие по результатам компьютерного моделирования (in silico [1]) молекулы — кандидаты в лекарства — могут стать «чудесной таблеткой», например, из-за их нерастворимости, стерических (пространственных) ограничений на связывание с молекулой-мишенью (обычно белком) или защиты опережающим патентом. Даже при выполнении этих условий потенциальное лекарство может провалить клинические испытания из-за проблем с доставкой к пораженным тканям и органам. Оно может слишком быстро разрушаться и выводиться из организма, так и не достигнув цели. Кросс-реактивность — еще одна проблема при создании лекарств. Она заключается в том, что молекулы потенциального лекарства часто могут взаимодействовать не только с предполагаемой нами мишенью, но и с другими белками в организме. В таком случае побочные эффекты могут быть даже более опасны, чем сама болезнь.
Есть множество причин, по которым какое-либо активное вещество может так и не стать лекарством. Яркий пример недостаточного исследования активного вещества перед выводом его на фармацевтический рынок — трагедия со снотворным средством талидомидом. Из-за массового использования талидомида женщинами во время беременности в Европе в период с 1956 по 1962 года родилось порядка восьми−двенадцати тысяч (по разным оценкам) детей с серьезными врожденными уродствами. По данным, собранным профессором В. Ленцем, около 40% детей, подвергшихся воздействию препарата на стадии развития плода, умерли еще до своего рождения. Дело в том, что молекула талидомида может существовать в виде двух оптических изомеров (право- и левовращающего). Оказалось, что один из них обеспечивает терапевтический эффект препарата, а другой является причиной его негативного воздействия на развитие плода (тератогенности) и других побочных эффектов. Таким образом, необходимо провести как можно более подробную проверку свойств вещества и его поведения в организме, прежде чем выпускать его на рынок. Далее мы поговорим подробнее о некоторых интересных методах, помогающие решить такую задачу: смена скелета (scaffold hopping), поиск по фармакофору и по молекулярному подобию.
Современная разработка лекарств и драг-дизайн
Драг-дизайн, или рациональное конструирование лекарств, — это активно развивающаяся в последнее время область исследований. Основы этого направления хорошо изложены на русском языке в статье Антона Чугунова «Драг-дизайн: как в современном мире создаются новые лекарства» [2]. Для многих болезней еще нет достаточно эффективных лекарств, путь молекулы-кандидата на фармацевтический рынок занимает десятки лет, и создание одного лекарства стоит несколько миллиардов долларов [3]. Базы данных (например, PubChem, ChEMBL, ZINC) содержат десятки миллионов химических соединений. Проверить экспериментально, является ли каждая молекула лекарством от какой-либо болезни — практически невыполнимая задача. Именно поэтому необходимо проводить первичный отбор в этом огромном пространстве химических соединений in silico, основываясь на их структурах и известных свойствах. Для этого есть много различных подходов, например, молекулярный докинг [4] и молекулярная динамика [5]. Молекулярный докинг — метод молекулярного моделирования, который позволяет предсказывать наилучшее положение лиганда (обычно, малой молекулы) относительно белка-мишени, используя их трехмерные структуры и оценочные функции энергии взаимодействия молекул. Молекулярная динамика основана на использовании уравнений движения атомов и эмпирических функций потенциальной энергии для расчетов межатомных взаимодействий и описания эволюции молекулярной системы во времени.
Первый пример успешного использования драг-дизайна, основанного на знании структур молекулы мишени и лигандов — ингибитор карбоангидразы дорзоламид (противоглаукомное средство), который был утвержден в качестве лекарства еще в 1995 году. Другой важный пример — ингибитор фермента тирозинкиназы иматиниб (противолейкозный препарат). Он был разработан методами рационального драг-дизайна — скрининга библиотек химических соединений для поиска вещества, ингибирующего определенный белок-мишень. Он является «таргетным лекарством», то есть губительно действующим только на раковые клетки. Его изобретатели получили премию Ласкера (которую иногда называют американской Нобелевской премией) в 2009 году и Премию Японии в 2012 году.
Выбор мишеней
Прежде, чем искать лекарство от какой-либо болезни, нужно понять молекулярные основы ее развития. Изучить причины и узнать, взаимодействия между какими белками являются ключевыми. Таким образом мы можем описать развитие заболевания на молекулярном уровне с помощью каскада реакций . Это необходимо, чтобы определить, какие белки являются наиболее перспективными мишенями (объектами, на которые нужно воздействовать, чтобы исправить или компенсировать нарушения, ставшие причиной заболевания).
Кстати, в современной системной биологии практика записи каскадов реакций и вообще иерархий биохимических и биологических процессов получает все большее распространение [6]. — Ред.
Что нужно учитывать при моделировании, или откуда берутся окольные тропы
Представим, что наши вычислительные мощности невероятно велики, и мы можем проверить, оценив энергию связывания, как взаимодействуют практически все известные малые молекулы с белком-мишенью (на самом деле это далеко не так). Пусть в результате такой проверки мы выбрали некоторое количество молекул, показавших лучшие результаты. Как вы думаете, все ли они могут стать лекарствами? Чтобы ответить на этот вопрос, не проводя экспериментов с клетками и, тем более, организмами, нам нужно провести дополнительную проверку in silico. Давайте вспомним, что малая молекула должна удовлетворять эмпирически полученному «правилу пяти» Липинского (иметь менее пяти атомов-доноров водородной связи; обладать молекулярным весом меньшим 500; иметь липофильность (log P — коэффициент распределения вещества на границе раздела вода-октанол) менее 5; иметь не более 10 атомов азота и кислорода) [2]. Нельзя также забывать и про метаболические характеристики (ADMET: всасывание, распределение, метаболизм, экскрецию, токсичность). Кроме всего этого, необходимо, чтобы молекула-кандидат была способна взаимодействовать в пространстве с активным центром мишени и удовлетворяла стерическим ограничениям. Может быть так, что потенциально активная молекула слишком большая и не может «пробраться» к активному сайту мишени, или она имеет сложную структуру, из-за чего ее очень трудно и дорого синтезировать. Другой вариант: молекула очень маленькая и «универсальная», — тогда она взаимодействует не только с нашей мишенью, но и со многими другими белками в организме человека. Это приводит к побочным эффектам, которые могут быть очень серьезными. Картина получается не самой радостной, а дорога лекарства на рынок все более запутанной. «С тех пор все тянутся передо мною кривые, глухие окольные тропы», — по словам Ёсано Акико, японской поэтессы. Хорошо, что есть способы, помогающие не запутаться в этих окольных тропах драг дизайна.
Смена скелета
Когда вы нашли молекулу-кандидат в лекарства, нужно провести оптимизацию, улучшить ее свойства, чтобы увеличить вероятность успеха при экспериментальной проверке. Рассматривая молекулу-кандидат (лиганд) мы можем выделить в ней важные для взаимодействия с белком-мишенью части, структурную основу — скелет — и другие группы, предположительно не участвующие в интересующем нас взаимодействии или даже мешающие контакту лиганда с мишенью. Смена скелета (scaffold hopping) нужна, когда мы хотим сохранить важные для взаимодействия с мишенью части лиганда и заменить остальные на более подходящие. Конечно же, необходимо при этом учитывать изменение энергии связывания лиганда с мишенью. Кроме этого, изменяются конформационные свойства лиганда, что сказывается на энтропии взаимодействия.

Рисунок 1. Принцип «смены скелета»: совпадение форм, поиск особых групп, определяющих свойства, сама смена скелета, поиск по подобию.

Рисунок 2. Исходная молекула и молекула — результат смены скелета
При смене скелета мы получаем много новых вариантов, среди которых выбираем лучшие, наиболее нам подходящие по трехмерной структуре, энтропии, энергии связывания с белком-мишенью и другим характеристикам. Смена скелета лиганда расширяет нам поле возможностей, дает множество вариаций. Как будто у нас есть набор фрагментов конструктора, и мы хотим собрать некоторую упорядоченную и стабильную структуру (используя эти фрагменты и некоторые связующие их), чтобы потом присоединить ее к чему-нибудь еще. Как будто мы потеряли один крайний кусочек паззла, но знаем, что должно быть в том месте, которым он присоединяется к картине, в результате чего делает ее завершенной. Таким образом, мы можем, например, из большого и трудносинтезируемого лиганда сделать маленький, хорошо способный пробираться к сайту связывания. Словно вырезать из картона недостающий кусочек паззла, отбросив все лишнее.
Есть еще одна интересная область применения смены скелета, связанная с так называемыми опережающими патентами. Такие патенты выдаются на некоторое время компаниям, утверждающим о наличии некоторой лекарственной активности у определенной молекулы. В течение этого времени они должны доказать, что эта активность действительно есть. А что же делать вам, если вы сами нашли активность у этой молекулы и хотите запатентовать ее? Когда вы делаете смену скелета, результатом является уже другая молекула, не закрытая патентом, но обладающая интересующей биологической активностью [7], [9–11].
Поиск похожих молекул с использованием фармакофоров
Фармакофор — это набор пространственных и электронных признаков, необходимых для обеспечения оптимальных взаимодействий с биологической мишенью, которые могут вызывать или блокировать её биологический ответ. Такими признаками могут быть гидрофобность, ароматичность, донорно-акцепторные свойства, заряд. Мы можем использовать поиск по фармакофору при виртуальном (компьютерном) скрининге больших баз данных, библиотек химических соединений. Особенно полезен такой подход, когда у нас недостаточно информации о строении мишени и набора лигандов для использования методов моделирования, основанных на использовании трехмерных структур.

Рисунок 3. Пример фармакофора молекулы — агониста серотонинового рецептора 5-HT2c

Рисунок 4. Пример трехмерной фармакофорной модели

Рисунок 5. Результаты поиска по молекулярному подобию
Поиск по молекулярному подобию
Структурно похожие молекулы обычно обладают сходными биологическими свойствами. В этом заключается принцип молекулярного подобия. Пусть мы знаем, что некоторый лиганд активно взаимодействует с мишенью, но в силу разных причин (взаимодействие с другими белками организма и токсичность, нарушение свойств ADMET, защита патентом) не может быть использован нами в качестве нового лекарства. Тогда мы можем осуществить поиск по подобию и найти подходящую молекулу-лиганд, которая впоследствии с большей вероятностью станет лекарством.
Заключение, или что нам учить, чтобы создать лекарство?
Над созданием лекарств трудятся многие лаборатории, фармацевтические компании и даже школьники. Об одной из работ школьников в этой области написана статья Веры Башмаковой «НИИ ЧАВО себе!» [14]. Для того, чтобы найти малую молекулу, обладающую лекарственной активностью против определенного заболевания, нужно хорошо разбираться в молекулярной биологии, химии, био- и хемоинформатике, физике, программировать, использовать различные базы данных. Знание молекулярной биологии нужно для выбора белка-мишени, играющего ключевую роль в развитии заболевания. Физика помогает понять взаимодействия между молекулой-лигандом и белком-мишенью. Благодаря программированию, био- и хемоинформатике у нас есть методы для компьютерного создания лекарств. Базы данных нужны для поиска в них малых молекул лигандов и структур белков-мишеней. Знание химии помогает понимать свойства молекул, предсказывать их поведение в организме.
Примером комплексного подхода в драг-дизайне является работа Бергмана с коллегами, которая называется SHOP: scaffold HOPping by GRID-based similarity searches [16]. В ней использованы методы смены скелета, поиска по подобию, молекулярного докинга и некоторые математические вычисления. В качестве мишеней для демонстрации метода были взяты тромбин, протеаза ВИЧ-1 и нейраминидаза (белок вирусной мембраны, обладающий ферментативной активностью; один из ключевых белков вируса гриппа и хорошая мишень для драг дизайна; поиск ее ингибиторов начался еще в 1966 году). В создании лекарств велика роль везения, случая и вероятности. Именно поэтому мы можем использовать более продвинутые методы и «окольные тропы», когда «угаданная» молекула-кандидат не идеальна. Мы видим, что можно ее улучшить, нам хочется чего-то большего, и мы можем этого достичь.
«Открытие — это как влюбиться и достичь вершины горы после тяжелого подъема...» — сказал Макс Фердинанд Перутц, английский биохимик, лауреат Нобелевской премии 1962 года [8].
Благодарности
Пете Власову, открывшему мне возможности заниматься драг дизайном; Наталье Киреевой, которая читает отличные лекции по хемоинформатике в МФТИ (ГУ); Феде Кондрашову и Динаре Усмановой, которые вдохновляют меня заниматься наукой.
Литература
- In vivo — in vitro — in silico;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Herper M. (2013). The cost of creating a new drug now $5 billion, pushing Big pharma to change. Forbes;
- Thomas Lengauer, Matthias Rarey. (1996). Computational methods for biomolecular docking. Current Opinion in Structural Biology. 6, 402-406;
- Молекулярная динамика биомолекул. Часть I. История полувековой давности;
- ACSN — глобальный атлас сигнальных путей. От молекулярной географии рака к новым информационным технологиям в биологии;
- Gerhard Hessler, Karl-Heinz Baringhaus. (2010). The scaffold hopping potential of pharmacophores. Drug Discovery Today: Technologies. 7, e263-e269;
- Perutz M.F. (1981). True Science. The London Review of Books. 5;
- Flavien Quintus, Olivier Sperandio, Julien Grynberg, Michel Petitjean, Pierre Tuffery. (2009). Ligand scaffold hopping combining 3D maximal substructure search and molecular similarity. BMC Bioinformatics. 10, 245;
- Hongmao Sun, Gregory Tawa, Anders Wallqvist. (2012). Classification of scaffold-hopping approaches. Drug Discovery Today. 17, 310-324;
- Hans-Joachim Böhm, Alexander Flohr, Martin Stahl. (2004). Scaffold hopping. Drug Discovery Today: Technologies. 1, 217-224;
- Deschênes A. and Sourial E. (2007). Ligand Scaffold Replacement using MOE Pharmacophore Tools. Chemical Computing Group Inc.;
- A.V. Koval, P. Vlasov, P. Shichkova, S. Khunderyakova, Y. Markov, et. al.. (2014). Anti-leprosy drug clofazimine inhibits growth of triple-negative breast cancer cells via inhibition of canonical Wnt signaling. Biochemical Pharmacology. 87, 571-578;
- НИИ ЧАВО себе!;
- Soma Mandal, Mee'nal Moudgil, Sanat K. Mandal. (2009). Rational drug design. European Journal of Pharmacology. 625, 90-100;
- Rikke Bergmann, Anna Linusson, Ismael Zamora. (2007). SHOP: Scaffold HOPping by GRID-Based Similarity Searches. J. Med. Chem.. 50, 2708-2717.



Комментарии
0Чтобы оставить комментарий, необходимо
войти