Белковые галлюцинации: как справляется AlphaFold?
18 ноября 2022
Белковые галлюцинации: как справляется AlphaFold?
- 2081
- 0
- 3
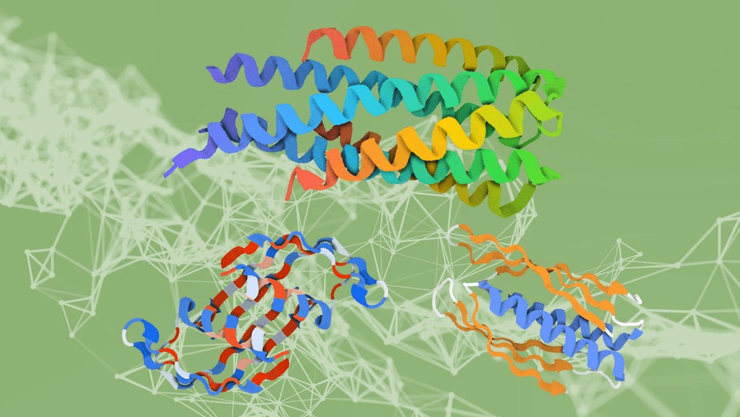
Олигомеры белков, успешно спроектированных нейросетью ProteinMPNN
-
Автор
-
Редакторы
-
Рецензент
Не успев пережить триумф нейросетей (в лице AlphaFold) в предсказании строения белков, структурная биоинформатика взялась за обратную задачу: подбор последовательности для заданной трехмерной укладки. Но решение этой проблемы пока сдерживается фундаментальными ограничениями, присущими и самой программе AlphaFold. В этой колонке я постараюсь объяснить, почему нейросети в структурной биоинформатике не стоит считать панацеей.
Практически с самого рождения «Биомолекулы» (а это аж 2007-й год!) одной из любимых тем портала было компьютерное моделирование структур белков и лежащие в его основе теоретические проблемы. Уже в 2008 году у нас вышла статья «Торжество компьютерных методов: предсказание строения белков» [2], а в 2016 году в конкурсе «Био/Мол/Текст» приняла участие публикация «Проблема фолдинга белка» [3]. Вкратце их обе можно пересказать так: последовательность белка однозначно определяет его трехмерную структуру, только вот вычислительный способ «перевода» одной структуры в другую до недавнего времени был неизвестен. Конечно, можно запустить симуляцию сворачивания белка «в лоб», но это слишком долгий процесс даже при наличии мощных суперкомпьютеров и распределенных вычислений (ну и, честно сказать, срабатывает он только для самых маленьких и «хорошо фолдированных» белков, да и то не точно: «Миллисекундный барьер взят!» [4]). Еще можно виртуально «натянуть» исследуемый белок на его «родственника» с известной структурой (это называется моделированием по гомологии) — но только если такой «родственник» есть.
Так как мы постоянно отслеживаем новые решения в этой области, вместе с научным миром мы были воодушевлены появлением в 2019–20 годах AlphaFold — нейросети, которая умеет предсказывать структуру белков. При создании этой сети разработчикам наконец удалось найти математическое решение проблемы «перевода» последовательности в трехмерную структуру: они обучали сеть на наборе дистанций между аминокислотными остатками и применяли метод градиентного спуска [5]. Конечно, это не четкая формула, тем более не основанная на какой-то понятной физике, но все же это рабочий подход для искусственного интеллекта.
Появление новой нейросети вызвало волну ажиотажа и энтузиазма в научном сообществе. Поначалу у многих возникло впечатление, что проблема фолдинга уходит в небытие. Но так ли это на самом деле? Спустя два года мы пытаемся подвести промежуточный итог и ответить на этот вопрос.
Проблема фолдинга решена?
Проблема фолдинга белка (читаем) — это невозможность компьютерным путем предсказать трехмерную структуру белка по его последовательности. Проблема довольно-таки досадна: в естественных условиях укладка жестко определяется последовательностью. Основные причины «бессилия» компьютеров — очень сложный характер изменения энергии белка при фолдинге в сочетании с большим количеством потенциально возможных конформаций (чтобы перебрать их все «в лоб», даже самому белку потребовалось бы больше времени, чем существует Вселенная — эта драматическая нестыковка даже получила название «парадокса Левинталя»).
Прямое моделирование методом молекулярной динамики, где можно было бы пронаблюдать процесс укладки белка в модели физических сил, оказывается технически ресурсоемким процессом: белок сворачивается за миллисекунды, а молекулярная динамика хорошего качества — это пока что лишь десятки-сотни микросекунд. Один из немногих опубликованных миллисекундных расчетов — пока исключение, но никак не правило.
По сути, AlphaFold не решает эту фундаментальную биофизическую проблему, ничего не добавляя к нашему знанию о сворачивании белков in vivo и in vitro — он ее элегантно обходит. Он не моделирует процесс свертывания белка, который происходит в естественных условиях. Первая версия AlphaFold просто предсказывала с помощью нейросети набор попарных расстояний между аминокислотными остатками по последовательности, а дальше приводила структуру белка к предсказанным расстояниям методом градиентного спуска (рис. 1) [4].

Рисунок 1А. Принцип работы AlphaFold: обучение нейросети. Здесь трехмерная структура каждого белка, на котором будет учиться нейросеть, преобразуется в матрицу расстояний между аминокислотными остатками, или дистограмму. На вход нейросети подаются дистограммы вместе с последовательностями белков, и сеть учится устанавливать соответствие между последовательностью и дистограммой.
рисунок создан с помощью BioRender.com

Рисунок 1Б. Принцип работы AlphaFold: предсказание. В «рабочем режиме» нейросеть принимает на вход последовательность белка и предсказывает по ней дистограмму.
рисунок создан с помощью BioRender.com

Рисунок 1В. Принцип работы AlphaFold: градиентный спуск. Структура белка «сворачивается» таким образом, чтобы приблизить ее дистограмму к предсказанной нейросетью на предыдущем этапе — математически для этого используется метод градиентного спуска.
рисунок создан с помощью BioRender.com
Вторая версия (AlphaFold 2) использует для предсказания структуры белков эволюционные данные. Даже если у белка нет эволюционных родственников с известной структурой, это не значит, что их нет вообще. Скорее наоборот — в базах данных полно гомологичных белков, для которых тоже не выяснена структура. Выровняв их друг с другом (построив множественное выравнивание), можно понять, как эволюционировали те или иные аминокислотные остатки в белке. И если, например, два остатка эволюционировали вместе, значит, они могут быть как-то связаны структурно — например, образовывать прямой контакт. Гораздо более сложная архитектура, чем в первой версии, позволяет буквально вычислить структуру по эволюции (рис. 2). Очень интересный с биологической точки зрения и многообещающий подход. Подробно принцип работы второй версии изложил Борис Бурков в своем блоге на Хабре.

Рисунок 2. Принцип работы AlphaFold 2. Здесь уже нет никакого градиентного спуска, зато добавилось использование эволюционных данных — множественных выравниваний.
[6], CC BY 4.0
До появления AlphaFold, конечно, «обходные» решения тоже были. Лидером среди них была программа Rosetta, но она полагалась на большее использование «грубой силы»: она генерирует сразу много конформаций белка, собирает их в похожие группы — кластеры — и отбрасывает самые слабозаселенные. Потом программа выбирает структуры с самой низкой энергией (рис. 3) [7]. Такой подход тоже помогает обойти прямое моделирование фолдинга и сделать задачу технически реализуемой — но за счет работы с большим количеством структур она оставалась все еще очень ресурсоемкой и требующей большого количества серверов. Более того, разработчики Rosetta даже использовали для ее решения краудсорсинг на базе распределенных вычислений — это проект Rosetta@Home [8]. В общем, предсказать структуру белка с приемлемыми точностью, стоимостью и временем было задачей трудновыполнимой. Оставалось разве что прибегать к грубым методам типа крупнозернистой молекулярной динамики, которые резко снижали точность.

Рисунок 3. Алгоритм Rosetta. Программа запускает моделирование параллельно во много потоков, результатом чего становится большой ансамбль структур. В идеале они все получились бы одинаковыми, но в реальности так никогда не бывает: из-за случайного характера сборки, все финальные конформации немного (или не немного) различаются. Некоторые запуски приведут к структурам, сильно отличающимся от большинства. Каждое моделирование воспроизводит фолдинг — а значит, вероятность того, что только одна сборка из многих привела к похожей на нативную конформацию, очень мала. Поэтому конформации из полученного набора с близкими структурами и значениями энергии объединяются в группы — кластеры. Если в кластере мало представителей — он отбрасывается. Среди оставшихся выбирают один с наименьшей средней энергией: нативная упаковка, скорее всего, именно там. Результатом работы Rosetta может быть любая структура из финальной группы — они все похожи друг на друга.
рисунок Васили Сулеймановой по [9]
Основная новизна AlphaFold — в скорости. За счет отсутствия опоры на вычислительную «грубую силу» он решает задачи предсказания структуры белка куда меньшими ресурсами без снижения точности. В сфере предсказания структур белка его появление было сходно со сменой огромных ЭВМ на компактные персональные компьютеры и ноутбуки: в целом прежний функционал стал дешевым, быстрым и удобным. Неудивительно, что на практике AlphaFold воспринимается как прорыв.
Именно так и называется премия, которой удостоились исследователи — Breakthrough Prize, или «Премия за прорыв» [10]. Премия составляет 3 миллиона долларов и является самой «денежной» наградой в науке. Можно ожидать в дальнейшем получение авторами и других премий — вплоть до Нобелевской. С вычислительной точки зрения это действительно революция.
Эта революция позволила к настоящему моменту предсказать структуры более чем для двухсот миллионов белков — все они находятся в открытом доступе. Однако ученые предупреждают, что это именно предсказания, а не экспериментально подтвержденные укладки — и относиться к ним надо соответствующим образом. Джули Форман-Кей, специалист по биофизике белка из Университета Торонто, замечает: «Сейчас есть ощущение, что людям больше не нужно определять структуру экспериментальными методами, но это не так» [11]. Ученые всерьез беспокоятся, что появление AlphaFold приведет к снижению финансирования и объемов подготовки специалистов по анализу структуры белка экспериментальными методами.
Но и это еще не конец. Помимо разных редакций самого AlphaFold, появляются и аналогичные нейросетевые алгоритмы: компания Meta (которая признана экстремистской и запрещена в России) в этом году обнародовала первые результаты испытаний своей нейросети для предсказания структур белков — ESMFold. Эта сеть уступает AlphaFold в точности, зато дает ему фору по части скорости — она смогла предсказать трехмерные структуры аж более чем для 600 миллионов (!) белков всего за две недели, добавив к и так бездонному множеству совсем уж «темную материю» гипотетических метагеномных белков. — Прим. авт.
У AlphaFold есть одна не очень приятная слабость — в основном у него получаются модели «сферических белков в вакууме»: он ничего не может сказать о том, как белок будет взаимодействовать с лигандами или даже с другими белками. Само решение задачи фолдинга белка таким методом не предполагает присутствия в моделируемой системе чего-либо, кроме аминокислотных остатков. Получаются модели белка «самого по себе», без каких-либо партнеров, с которыми он в нормальных условиях взаимодействует.
При этом каждое взаимодействие меняет конформацию белка, иногда весьма драматически — в итоге получается, что AlphaFold не может нам сказать, как себя поведет белок в действии. Если бы мы были инопланетянами, раздобыли ген белка глобина земных организмов и попытались бы его «свернуть» AlphaFold'ом, смогли бы мы вообще понять, что к нему должен «прилагаться» гем? Смогли бы отмоделировать «кармашки» для его связывания таким образом, чтобы его туда можно было поместить методом докинга? Сложный вопрос, на который мы рискуем так и не получить ответа — наш AlphaFold заведомо «знает», как устроены глобины. Но у него нет таких «знаний» о других белках — включая ферменты, которые он моделирует. Возможное техническое решение для такой задачи пока находится только на стадии препринта [12], и насколько оно покажет себя на практике — пока неясно.
И это может затруднить использование AlphaFold для создания новых белков.
Что? Создание новых белков?
Разделавшись с проблемой фолдинга белка, ученые взялись за обратную задачу — дизайн аминокислотных последовательностей, которые бы «сворачивались» в заданную трехмерную структуру. Однако единственное, чему удалось «научить» AlphaFold, помимо «просто» предсказания белковых структур, — это восстановление четвертичных структур, состоящих из нескольких или многих субъединиц. Соответственно, когда пришла пора «придумывать» новые последовательности белков, такая постановка задачи оказалась для искусственного интеллекта потолком, который не так просто преодолеть.
Прежде всего, сам по себе AlphaFold оказался для этого практически бесполезным. Команда Дэвида Бейкера — разработчика программы Rosetta и директора Института белкового дизайна в Калифорнии — пыталась приспособить AlphaFold для дизайна белков. Их метод, названный белковой галлюцинацией (hallucination), позволил создать «с нуля» несколько небольших белков [13]. Но попытка дизайна более крупных агрегатов заданной формы потерпела неудачу при первой же экспериментальной проверке. Предсказания не только не оправдались — белки вообще не образовывали упорядоченных структур. «Они вообще не сворачивались: просто лежали грязью на дне пробирки», — говорит Бейкер [14].
Другая подгруппа в лаборатории Бейкера создала свою нейросеть, названную ProteinMPNN и не основанную на AlphaFold непосредственно. Ее научили «придумывать» последовательности, при фолдинге формирующие крупные белковые наночастицы из множества субъединиц (рис. 4). Эти структуры напоминают по форме цилиндры или колёса и ранее не существовали в природе. Во-первых, это красиво! Но во-вторых, как-то не очень полезно…

Рисунок 4. Трехмерные структуры белков, для которых искусственный интеллект смог подобрать последовательность. Такие структуры не встречаются в природе.
Ian C Haydon / UW Institute for Protein Design / Nature
Для белка главное — не иметь вычурную форму (в конце концов, ее нельзя увидеть глазами), а делать что-то полезное: связывать токсин или расщеплять какое-нибудь вещество. То есть наш «заказ» искусственному интеллекту должен включать в себя определенное строение связывающего участка. Пока решения на базе нейросетей для такого «каприза» только на стадии разработки [14]. Причина уже упоминалась — само решение задачи фолдинга белка с помощью нейросети, работающей с дистограммами или множественными выравниваниями, не учитывает всех взаимодействий. Это лишняя переменная, которой нужно исхитриться найти место в такой постановке вопроса.
Зато программа Rosetta с такой задачей ранее справлялась: она создавала белок-антидот к сердечному гликозиду диоксигенину, фермент для расщепления глиадина (может пригодиться людям с целиакией), термостабильные версии известных белков и много чего еще — «Биомолекула»: «Конструкторское бюро белков» [7].
Создание таких белков начинается с дизайна связывающего участка — он «отрисовывается» согласно структуре лиганда, чтобы подходить к ней лучшим образом. Поэтому на момент запуска самой программы предсказания расположение аминокислот связывающего участка уже задано — но для Rosetta это не проблема. Алгоритму на основе энергии и кластеризации ничего не мешает аккуратно «обернуть» этот участок оставшейся частью белка и при этом не изменить структуру активного центра (рис. 5) [7].

Рисунок 5. Чтобы спроектировать белок под определенный лиганд, сначала создается окружение лиганда — его связывающий участок. Потом программа Rosetta «проектирует» весь остальной белок.
рисунок Васили Сулеймановой
Лаборатория Дэвида Бейкера уже взяла этот факт на вооружение и предложила немного парадоксальное решение: скрестить ежа и ужа… то есть AlphaFold и Rosetta [15]! В другом препринте они же скрестили ProteinMPNN с Rosetta [16]. В общем, как оказалось, старые решения еще могут пригодиться, и их рано списывать со счетов.
Точно так же, рано списывать со счетов и экспериментальные методы определения структуры белков. К сожалению, здесь появляется «бутылочное горлышко» — не все структурные биоинформатики имеют доступ к необходимому экспериментальному оборудованию. Как бы мы ни старались совершенствовать компьютерные методы — мы по-прежнему заложники экспериментальных методик.
Заключение
В одной из недавних дискуссий в социальной сети, где я писал о своей работе по моделированию взаимодействия лейкотриенового рецептора с лигандами, мне был задан парадоксальный вопрос: «А зачем всё это делать, если AlphaFold уже всё свернул?» Подобные вопросы типичны в последнее время, и они отражают непонимание ограничений моделирования белков нейросетями. Существующие нейросети пока только учатся предсказывать поведение белка в динамике, изменения его формы при связывании с другим белком и связывание им малых молекул. Так что отказываться от других направлений структурной биоинформатики, считая AlphaFold панацеей, явно не стоит. Вдобавок самые эффективные и интересные разработки тоже могут ошибаться — так что экспериментальная проверка остается необходимой. В общем, AlphaFold и его версии — это новое слово в молекулярном моделировании. Но далеко не последнее.
Литература
- B. I. M. Wicky, L. F. Milles, A. Courbet, R. J. Ragotte, J. Dauparas, et. al.. (2022). Hallucinating symmetric protein assemblies. Science. 378, 56-61;
- Торжество компьютерных методов: предсказание строения белков;
- Проблема фолдинга белка;
- Миллисекундный барьер взят!;
- AlphaFold: нейросеть для предсказания структуры белков от британских ученых;
- John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, et. al.. (2021). Highly accurate protein structure prediction with AlphaFold. Nature. 596, 583-589;
- Конструкторское бюро белков;
- Тетрис XXI века;
- P. Bradley. (2005). Toward High-Resolution de Novo Structure Prediction for Small Proteins. Science. 309, 1868-1871;
- Zeeya Merali. (2022). AlphaFold developers win US$3-million Breakthrough Prize. Nature. 609, 889-889;
- Saey T.H. (2022). Has AlphaFold actually solved biology’s protein-folding problem? ScienceNews;
- Hekkelman M.L., de Vries I., Joosten R.P., Perrakis A. (2021). AlphaFill: enriching the AlphaFold models with ligands and co-factors. bioRxiv;
- Ivan Anishchenko, Samuel J. Pellock, Tamuka M. Chidyausiku, Theresa A. Ramelot, Sergey Ovchinnikov, et. al.. (2021). De novo protein design by deep network hallucination. Nature. 600, 547-552;
- Ewen Callaway. (2022). Scientists are using AI to dream up revolutionary new proteins. Nature. 609, 661-662;
- Wang J., Lisanza S., Juergens D., Tischer D., Anishchenko I., et. al. (2021). Deep learning methods for designing proteins scaffolding functional sites. bioRxiv;
- Bennett N., Coventry B., Goreshnik I., Huang B., Allen A., et. al. (2022). Improving de novo Protein Binder Design with Deep Learning. bioRxiv;
- Mehmet Akdel, Douglas E. V. Pires, Eduard Porta Pardo, Jürgen Jänes, Arthur O. Zalevsky, et. al.. (2022). A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol. 29, 1056-1067.



Комментарии
0Чтобы оставить комментарий, необходимо
войти