AlphaFold: нейросеть для предсказания структуры белков от британских ученых
04 августа 2020
AlphaFold: нейросеть для предсказания структуры белков от британских ученых
- 3534
- 0
- 8
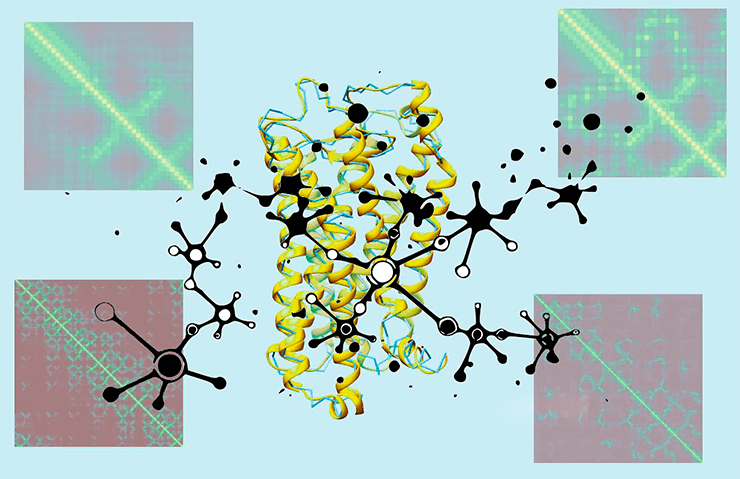
Нейронная сеть предсказывает структуру белка с использованием диаграмм дистанции, или дистограмм
коллаж автора статьи
-
Автор
-
Редакторы
Статья на конкурс «Био/Мол/Текст»: Уже не одно десятилетие биоинформатики бьются над проблемой предсказания структуры белков «с нуля». Решить проблему «в лоб», смоделировав процесс укладки, не особо получается — слишком малы вычислительные возможности современных компьютеров. Исследователи из Лондона предложили новое решение проблемы — теперь структуру белка предсказывает нейронная сеть! В профильном конкурсе искусственный интеллект обошел всех конкурентов и занял первое место. Как ему это удалось — читайте в нашей заметке!

Конкурс «Био/Мол/Текст»-2020/2021
Эта работа опубликована в номинации «Свободная тема» конкурса «Био/Мол/Текст»-2020/2021.
Генеральный партнер конкурса — ежегодная биотехнологическая конференция BiotechClub, организованная международной инновационной биотехнологической компанией BIOCAD.

Спонсор конкурса — компания SkyGen: передовой дистрибьютор продукции для life science на российском рынке.
Спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
«Книжный» спонсор конкурса — «Альпина нон-фикшн»
Черные дыры, суперкомпьютеры и видеоигры
Предсказание пространственной структуры белков — важнейшее назначение современной структурной биоинформатики [1]. И причины тому две. Во-первых, спрос на такие предсказания чрезвычайно высок. С появлением технологий NGS (секвенирования нового поколения) число доступных белковых последовательностей растет подобно снежному кому, а вот расшифровка пространственных структур до сих пор не поставлена на поток. Используемые для нее методики не только весьма сложны и дорогостоящи: для многих из них нет универсального «рецепта» эксперимента, как, например, для рентгеновской кристаллографии. Условия роста кристалла зачастую приходится подбирать почти интуитивно — и это мало способствует ускорению эксперимента. А драг-дизайнерам необходимы пространственные структуры в огромных количествах, чтобы разрабатывать лекарства против новых мишеней. Выход один — предсказывать.
А во-вторых и в главных, возможности биоинформатики тут резко ограничены. Относительно «потоковой» технологией является лишь сопоставительное моделирование, чаще всего реализуемое в виде моделирования по гомологии. Оно базируется на простой закономерности: гомологичные (эволюционно родственные и сходные по последовательности) белки в подавляющем большинстве случаев имеют почти одинаковую «укладку». Поэтому можно просто «натянуть» последовательность белка с неизвестной структурой на гомолог, для которого структура уже определена [2] (как мы сделали на рис. 1).

Рисунок 1а. Моделирование тромбоксанового рецептора мыши по тромбоксановому рецептору человека (структура PDB 6iiu). Шаблон — тромбоксановый рецептор человека — показан оранжевым, цепочка тромбоксанового рецептора мыши показана голубой линией. В помощью сервера Swiss-Model она «натянута» на рецептор человека так, чтобы совпадающие аминокислотные остатки совместились. Так и выглядит результат моделирования по гомологии.
собственная модель автора новости, визуализация в UCSF Chimera

Рисунок 1б. Модели по гомологии всегда нуждаются в уточнении — ведь для белка с другой первичной структурой конформация его «родственника» не будет энергетически выгодной. Для этого существуют готовые протоколы молекулярной динамики, зачастую выполненные в виде онлайн-ресурсов. Здесь представлен результат уточнения нашей голубой модели мышиного рецептора одним из таких ресурсов — GalaxyRefine (пурпурная лента — уточненная модель [3]).
Но такой метод абсолютно бессилен, если нет подходящего шаблона: у белка нет «родственников» с расшифрованной трехмерной структурой, у нас на руках последовательность мутантного белка с нарушенной укладкой (misfolding) или нужно решить задачу белковой инженерии — например, сконструировать «под ключ» фермент, который умел бы расщеплять, скажем, полиэтилен. Во всех этих случаях необходимы методы предсказания структуры белка «с нуля», или, как говорят биоинформатики, ab initio (лат. «с начала»). Как раз эта область остается одной из самых проблемных.
Ее проблемы и возможные решения ранее были подробно описаны на «Биомолекуле»: в одной из первых статей на сайте дан подробный обзор проблемы [2], а в 2016 году выпущен новый обзор [4]. В 2007 году рассказано о новых успехах на этом поприще [5]. Здесь повторю все вкратце.
Еще в середине XX века американский биохимик Кристиан Анфинсен сделал важное открытие: трехмерная структура белка определяется самой его последовательностью, и никакой дополнительной информации для сворачивания белка не требуется. В его опытах денатурированный фермент рибонуклеаза мог снова «сворачиваться» в свою активную конформацию и резать РНК дальше как ни в чем не бывало. Значит ли это, что трехмерная структура белка закодирована в самой последовательности его аминокислот? Можно сказать и так, но «взломать» этот код современной биоинформатике пока не по зубам.
Если ДНК, РНК и аминокислотная последовательность действительно связаны между собой однозначным соответствием (почему мы и можем предсказать последовательности всех белков организма по его геному), то при переходе от одномерной к трехмерной структуре белка в дело вступает довольно сложная физика.
Длинная полипептидная цепочка белка самопроизвольно сворачивается в «клубок» с наименьшей потенциальной энергией. В свою очередь, именно от последовательности аминокислотных остатков зависит, какая энергия будет наименьшей. Казалось бы, пока все просто.
Биоинформатики и хемоинформатики любят представлять все свои числа в виде гипотетического ландшафта в многомерном пространстве. Потенциальная энергия белка тоже образует такой ландшафт. Здесь каждому измерению будет соответствовать координата или угол поворота каждого аминокислотного остатка, и еще одно измерение отображает эту самую энергию. Нет-нет, не пытайтесь это представить, с многомерными пространствами работают, не представляя их интуитивно! Чтобы получилась наглядная картинка, придется «схлопнуть» все пространство координат в двухмерную плоскость. Тогда можно схематично нарисовать полученный ландшафт, как на рисунке 2. Видно, что ландшафт потенциальной энергии белка имеет форму черной дыры [6].

Рисунок 2а. Ландшафт потенциальной энергии белка, похожий на черную дыру
[6], рисунок адаптирован

Рисунок 2б. Аналогичная «черная дыра» в разрезе лучше показывает все неровности. Кроме того, у этой «дыры» есть «двойное дно» — образование аномальных амилоидо-подобных агрегатов.
[6], рисунок адаптирован
Вместо сингулярности у этой «черной дыры» — вполне себе конкретное «дно», соответствующее нативной конформации белка. Но, в отличие от астрономических черных дыр, сама «дыра» — не гладкая. Ее поверхность испещрена энергетическими барьерами и локальными минимумами энергии — «горами» и «впадинами». Белок при свертывании проходит по «тропинкам» между «горами» или вовсе перелезает через низкие холмы. Но как «горы», так и «впадины» гораздо больше, чем на рисунке, а вот по величине энергии отличаются друг от друга незначительно — то есть высокие пики соседствуют с глубокими пропастями [7]. А ведь речь не о двухмерном ландшафте, а подчас о пространстве с головокружительной мерностью — в зависимости от длины белка. Добавим к этому чисто техническую сложность: силовые поля, описывающие взаимодействия молекул в компьютерной модели, являются лишь приближениями реальных сил — для стопроцентной точности пришлось бы использовать уравнения квантовой механики, а это пока никакому суперкомпьютеру не под силу. Получается, что на существующих сейчас компьютерах нельзя просчитать тропинки, ведущие на дно «черной дыры» — программа их просто не увидит на обрывистом горном ландшафте. Так что...

Есть молекулярная динамика, где просто симулируется движение молекул по законам Ньютона с расчетом межмолекулярных и межатомных сил теми же самыми силовыми полями — при этом не ставится никаких целевых показателей в виде снижения энергии. Задаем начальные условия и оставляем нашу систему в виртуальном мире — а дальше смотрим, что получится. Пока это самый точный метод — но лишь за счет того, что симуляция сама по себе наиболее сложна и «реалистична» — как в компьютерных играх с хорошим игровым миром. Поэтому молекулярная динамика требует очень мощных компьютеров — и даже на них считается долго. И опять все упирается в имеющиеся у человечества вычислительные мощности: до недавнего времени можно было симулировать процессы, длящиеся в течение наносекунд, то есть миллиардных долей секунды. А фолдинг белка длится микросекунды, то есть тысячные доли. Между доступным и необходимым временем симуляции получается разрыв в 100–1000 раз. В 2010 году на «Биомолекуле» гордо отрапортовали, что миллисекундный барьер взят [8], и теперь стали возможны вычисления более 1 миллисекунды. Но все равно — пока мы можем «сворачивать» только очень короткие белки, и даже те имеют свойство надолго застревать в какой-нибудь «потенциальной яме» далеко от нативной конформации (видео 1).
Видео 1. Фолдинг фрагмента белка NTL9 (активатора транскрипции, вовлеченного в иммунитет растений), включающего первые 39 аминокислот. Моделирование методом молекулярной динамики. Время свертывания этого белка составляет примерно 1,5 мс, что лишь чуть-чуть превышает «миллисекундный барьер» [9]. И тем не менее такой фолдинг потребовал распределенных вычислений по проекту Folging@home, а сам белок надолго застрял в ненативной конформации — в «потенциальной яме». И, хотя этот фолдинговый триллер завершился удачно, так везет далеко не при каждой попытке «свернуть» белок методом молекулярной динамики.
Чтобы «облегчить» молекулярную динамику, ученые идут на различные ухищрения. Например, используют крупнозернистое (coarse-grained) представление молекул [10]. В ней целые аминокислотные остатки (или часто встречающиеся группы атомов) рассматриваются как цельные жесткие фигуры, своеобразные «псевдоатомы», для которых задаются потенциалы взаимодействия с другими компонентами системы (рис. 3).

Рисунок 3а. Различные способы крупнозернистого (coarse-grained) представления белков. Вверху слева — модель со всеми атомами, а на остальных изображениях — различные модели «огрубления», от довольно мягкой Rosetta CEN до радикальной SICHO.

Рисунок 3б. Одно из преимуществ такого подхода — сглаживание энергетических пиков и расщелин, что снижает вероятность «застревания» симуляции в ненативной конформации.
Такой подход значительно «облегчает» симуляцию и удлиняет доступное время, но при этом снижает точность, поэтому он пока не стал основной доступной и общепризнанной программой для предсказания структуры белков. Очевидно, что нужна какая-то новая технология...
В 2008 году известный «корифей» структурной биологии белков Дэвид Бэйкер совместно с профессорами информатики и инженерии Зораном Поповицем и Дэвидом Салесином решили превратить предсказание структуры белков... в увлекательную игру! Эта игра, в которой участники «сворачивают» белок, подробно описана в статье «Тетрис XXI века» [11]. Чем ниже расчетная потенциальная энергия полученной конформации, тем выше очки участника. Полученные модели потом используются для научных расчетов. Игра спроектирована таким образом, чтобы в нее могли играть неспециалисты — они и составляют львиную долю игроков. Такой вот игровой краудсорсинг. Основная идея игры состоит в том, что, хоть компьютер не может рассчитать правильную конформацию, ее вполне может почувствовать мозг человека. Стоп! Мозг! А что, если... Правильно! Нужен искусственный интеллект!
Дистанция лучше контакта! И лучше энергии!
Такую разработку представила лондонская компания DeepMind, занимающаяся искусственным интеллектом [12–14]. Вместе с ней в работе принимали участие Институт Фрэнсиса Крика (на российский манер он назывался бы НИИ биомедицины им. Фрэнсиса Крика) и Университетский колледж Лондона. Их основная идея — использовать в предсказании структуры белка нейронную сеть. Большое преимущество нейронных сетей — они способны обучаться, поэтому хорошо подходят для задач, где трудно представить алгоритм. В том числе — для предсказания структуры белка.
Британские ученые (ну не смейтесь! здесь все серьезно!) использовали сверточные нейронные сети (англ. convolutional neural networks). Они имитируют одну из самых сложно устроенных и вместе с тем одну из самых изученных нейронных систем мозга человека и животных — зрительные центры. Поэтому первоначально такие сети использовались в компьютерном зрении и распознавании изображений. На видео 2 наглядно показано, как такая сеть работает с изображением: многократно пропускает по нему маленький фильтр, «вылавливающий» какие-то паттерны. Примерно так же работает наша зрительная система. Математически такой алгоритм реализуется при помощи операции, называемой сверткой — отсюда и название.
Видео 2. Принцип обработки изображения сверточной нейросетью
Этот же алгоритм используется во всех других применениях сверточных сетей — в том числе и в работе британских ученых. Достаточно мысленно заменить экран с буквой «А» (на видео 2) на большой массив входных данных... а кстати, что британские ученые «скормили» нейросети? Ведь мало придумать нейросеть — надо решить, что подавать на вход!
Энергия свертывания белка, про которую я так много писал выше, определяется контактами между аминокислотами. Именно от них зависит образование слабых взаимодействий — водородных связей, гидрофобных контактов, солевых мостиков, стэкинга и тому подобных сил [15].
Напрашивается идея научить нейросеть предсказывать контакты, но исследователи заметили, что сеть проявляет куда бóльшие успехи, если учить ее предсказывать дистанции и углы! В итоге от контактов отказались, и все дальнейшее предсказание строилось на дистанциях и углах между аминокислотными остатками в трехмерной структуре.
Тренируя сверточные нейросети на известных структурах, ученые добивались, чтобы они точно предсказывали для новых белков распределение дистанций между аминокислотными остатками. И сети отлично справлялись со своей задачей. Получались своеобразные матрицы (рис. 4) — они и служили своеобразным оптимумом, к которому нужно стремиться при фолдинге. А для самогó сворачивания белка использовалась обычная для современного этапа математика — методы имитации отжига и градиентного спуска, широко используемые при решении задач на оптимизацию — не только применительно к белкам (рис. 5).

Рисунок 4. Результаты работы системы AlphaFold для трех белков. Верхний ряд — определенное экспериментально распределение дистанций. Каждая картинка включает в себя последовательность белка по горизонтальной и вертикальной осям (для экономии места не показаны), а сама дистанция обозначается яркостью: чем ярче пиксель на пересечении перпендикуляров от двух остатков, тем ближе эти остатки друг к другу. Очень яркая диагональ отражает тот факт, что каждый аминокислотный остаток «ближе всего» к самому себе. Такая иллюстрация называется дистограммой — от слов «дистанция» и «гистограмма». Средний ряд — дистограммы для того же белка, предсказанные AlphaFold’ом. Обратите внимание на сходство экспериментально определенных и предсказанных дистограмм. Нижний ряд — более привычное сравнение тех же моделей: белок, «свернутый» AlphaFold’ом (голубой) и экспериментально определенная модель того же белка (зеленая). И вновь — обратите внимание на сходство!

Рисунок 5. Фолдинг белка методом градиентного спуска. После предсказания распределения дистанций между остатками белок «сворачивается» методом градиентого спуска. Здесь этот процесс показан наглядно. Сравните с видео 1 — метод градиентного спуска дает более «гладкую» картину фолдинга без застревания в ненативных конформациях.
Исследователи фактически разрубили гордиев узел: полностью ушли от энергии, контактов, моделирования физических сил (все равно пока этого не умеем), а вместо этого занялись оптимизацией распределений дистанций (которые хорошо предсказываются методами машинного обучения). Сам по себе процесс «сворачивания» белка устроен куда проще, поэтому защищен от неприятных сюрпризов — например, застревания в энергетических ямах, как в молекулярной динамике (рис. 5). Но получилась ли методика точной? О да!
Проблема предсказания структуры белков — настолько сложная и объемная, что по ней есть отдельный конкурс. Называется он CASP — Critical Assessment of Protein Structure Prediction, или «критическая оценка предсказания белковых структур». Участникам предлагается предсказать структуру белков, еще не выложенных в открытый доступ. Конкурс проводится раз в два года. Разработчики AlphaFold участвовали в 13-м конкурсе (CASP13) и заняли первое место. А организаторы конкурса так описали их работу [14]:
Беспрецедентный прогресс возможностей компьютерных методов в предсказании структуры белков.
Любопытно, что AlphaFold оказался способен состязаться в точности не только с другими методами моделирования ab initio, но и с широко используемыми сейчас методиками моделирования по гомологии! Серьезный запрос, чтобы произвести настоящую революцию и прийти им на смену.
Уже сейчас научное сообщество не на шутку заинтересовалось разработкой: не успел выйти сам AlphaFold, как группа исследователей из частного Университета Бригама Янга в штате Юта (США) сделала свободный аналог! Я считаю, что это лучший комплимент компании DeepMind, хоть экономически немного невыгодный. Пока разработка описана только в препринте на bioRxiv [16].
Почти одновременно с публикацией статей по AlphaFold мир охватила эпидемия нового коронавируса — и проблема предсказания структуры белков встала как никогда остро: точные предсказания структуры вирусных белков потребовались срочно — чтобы дать миру шанс разработать эффективные лекарства. У AlphaFold появился шанс испытать себя в бою... и это испытание он с честью выдержал! Структура S-белка вируса, который образует «шипы» оболочки и обеспечивает связывание с клеткой-мишенью, была предсказана им точно — это выяснилось после публикации экспериментально определенной структуры S-белка в PDB. Впрочем, эти результаты авторы пока описали только в блоге компании, а какие-то выводы можно делать только после публикации в рецензируемых журналах. Но острота ситуации диктует необходимость такой предварительной публикации — тем более что в ней авторы поделились ссылкой на полученные ими модели вирусных белков (доступны все сразу в одном ZIP-архиве). Эти модели может использовать любой нуждающийся.
Так что искусственный интеллект в лице DeepMind уже включился в борьбу с пандемией. С чем еще он поможет нам справиться? Время покажет!
Литература
- 12 методов в картинках: «сухая» биология;
- Торжество компьютерных методов: предсказание строения белков;
- Lim Heo, Hahnbeom Park, Chaok Seok. (2013). GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Research. 41, W384-W388;
- Проблема фолдинга белка;
- Новые успехи в предсказании пространственной структуры белков;
- Bob Schiffrin, David J. Brockwell, Sheena E. Radford. (2017). Outer membrane protein folding from an energy landscape perspective. BMC Biol. 15;
- Нолтинг Б. Новейшие методы исследования биосистем. М.: «Техносфера», 2005. — 256 с.;
- Миллисекундный барьер взят!;
- Vincent A. Voelz, Gregory R. Bowman, Kyle Beauchamp, Vijay S. Pande. (2010). Molecular Simulation ofab InitioProtein Folding for a Millisecond Folder NTL9(1−39). J. Am. Chem. Soc.. 132, 1526-1528;
- Sebastian Kmiecik, Dominik Gront, Michal Kolinski, Lukasz Wieteska, Aleksandra Elzbieta Dawid, Andrzej Kolinski. (2016). Coarse-Grained Protein Models and Their Applications. Chem. Rev.. 116, 7898-7936;
- Тетрис XXI века;
- Andrew W. Senior, Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, et. al.. (2020). Improved protein structure prediction using potentials from deep learning. Nature. 577, 706-710;
- Andrew W. Senior, Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, et. al.. (2019). Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins. 87, 1141-1148;
- Senior A., Jumper J., Hassabis D., Kohli P. (2020). AlphaFold: using AI for scientific discovery. DeepMind;
- Роль слабых взаимодействий в биополимерах;
- Wendy M. Billings, Bryce Hedelius, Todd Millecam, David Wingate, Dennis Della Corte. (2020). ProSPr: Democratized Implementation of Alphafold Protein Distance Prediction Network. Cold Spring Harbor Laboratory;
- Компьютерные технологии против коронавируса: первые результаты.



Комментарии
0Чтобы оставить комментарий, необходимо
войти