Ингибиторы Ras: в поисках Грааля таргетной терапии
08 октября 2021
Ингибиторы Ras: в поисках Грааля таргетной терапии
- 2707
- 0
- 5
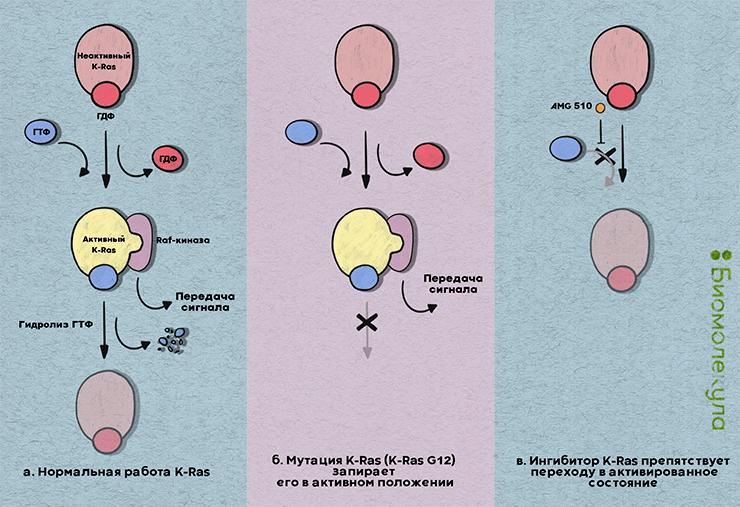
Как инактивировать K-Ras? а — Нормальный цикл K-Ras: ГТФ замещает ГДФ, активируя K-Ras и его мишени, гидролиз ГТФ деактивирует белок. б — Мутация K-Ras запирает его в активном положении. в — Ингибитор K-Ras препятствует переходу в активированное состояние.
-
Автор
-
Редакторы
-
Иллюстратор
K-Ras, один из первых найденных онкогенов, десятилетиями оставался недостижимым для лекарств. Неприступная мишень таргетной терапии была покорена благодаря новым подходам к скринингу, тщательной работе медицинских химиков и структурных биологов. Развитие этого направления может открыть дорогу к лечению одной из самых смертельных опухолей — рака поджелудочной железы.

Современные лекарства
Спецпроект о современных лекарствах, истории их создания, методах разработки и тенденциях развития.
Партнер спецпроекта — компания Cytiva — образовалась в результате продажи подразделения GE Healthcare Biopharma корпорации Danaher Corporation. Cytiva — глобальный поставщик технологий и услуг, которые продвигают и ускоряют разработку и производство терапевтических средств. У компании богатое наследие, насчитывающее сотни лет. Клиенты Cytiva проводят мероприятия по спасению жизни, начиная от фундаментальных биологических исследований и заканчивая разработкой инновационных вакцин, биологических препаратов и новейших клеточных и генных терапий. Задача компании — предоставить инструменты и услуги, которые им необходимы, чтобы они работали лучше, быстрее и безопаснее, что приведет к лучшим результатам для пациентов.
В 1970-х годах исследователи начали подбираться к механизмам развития опухолей. Выделилось два подхода: исследование вирусов, трансформирующих нормальные клетки в опухолевые (в частности, вызывающих опухоли у животных), и новый метод трансфекции ДНК, позволяющий перенести генетический материал из опухолевых клеток в нормальные, что приводит к трансформации. Работая с вирусами, ученые поняли, что они несут аналоги клеточных генов, захваченных ими в прошлом, и что эти гены присутствуют в норме у всех многоклеточных организмов. Двумя такими вирусами были вирус саркомы мышей Харви и вирус саркомы мышей Кирстена, полученные пассированием ретровирусов саркомы мышей (MLV) в крысах.
Было очевидно, что эти вирусы захватили какие-то гены из генома крыс: и действительно, гибридизация РНК этих вирусов показала наличие исходного клеточного протоонкогена в геноме крыс и других организмов. Параллельно в других исследованиях трансфекция ДНК из мышиной опухоли мочевого пузыря, полученной с помощью химического канцерогена, показала, что только ДНК из мутированных клеток может трансформировать нормальные клетки. Применение зарождающегося тогда секвенирования показало, что ген, захваченный вирусом саркомы Харви, гомологичен клеточному онкогену, мутация которого вызывала опухоль. Ген назвали h-ras (Harvey rat sarcoma), а его гомолог из вируса саркомы Кирстена k-ras (Kirsten rat sarcoma) [1]. Вскоре обнаружили и третий ген этого семейства — n-ras.
Белки семейства Ras оказались одним из центральных компонентов передачи сигналов от рецепторов роста. В отличие от большинства компонентов этих сигнальных каскадов, Ras — не киназы, а ГТФазы. Они могут находиться в двух состояниях: активном (связанном с ГТФ) и неактивном (связанным с ГДФ), переход между которыми требует белков еще двух классов (GAPов и GEFов; см. рис. 1). Белки Ras состоят из двух функциональных доменов: G-домена, который связывает ГТФ, и мембран-связывающего домена (рис. 1) [2]. Получив сигнал от рецептора, K-Ras, как диспетчер, координирует активацию более 80 сигнальных путей, прежде всего митоген-активируемого протеинкиназного сигнального каскада (MAPK) и сигнального пути PI3K/AKT.

Рисунок 1. Переход RAS-GDP (неактивный) ⇋ RAS-GTP (активный) происходит с помощью двух классов белков. Одни называются факторами обмена гуаниновых нуклеотидов (GEFs) и способствуют обмену ГДФ на ГТФ, а другие — ГТФаза-активирующие белки (GAPs) — выполняют обратную функцию и помогают катализировать гидролиз ГТФ до ГДФ, инактивируя Ras. Включенный Ras активирует свои мишени, прежде всего киназы Raf и PI3K.
Подавляющее большинство мутаций K-Ras меняет лишь две аминокислоты: 12 и 13 глицины, с более редкими мутациями глутамина 61 (рис. 2). Эти изменения оставляют белок в активном состоянии, в котором он связан с ГТФ, и препятствуют гидролизу ГТФ→ГДФ. Мутации G12 и G13 стерически препятствуют связыванию GAP-белков, помогающих K-Ras катализировать гидролиз ГТФ и перейти в неактивную форму, в то время как мутации Q61 препятствуют координации с молекулой воды, что ингибирует гидролиз ГТФ. Всего лишь одного такого изменения достаточно, чтобы перестроить множество процессов — подчиняясь указаниям (слегка уже неадекватного) диспетчера, клетка начинает бесконтрольно делиться, перестраивает свой метаболизм и настраивается на миграцию (рис. 3).

Рисунок 2. Частота мутаций K-Ras: преимущественно заменяются глицины 12 и 13, а также глутамин 61.

Рисунок 3а. В здоровых клетках K-Ras выполняет важные функции, контролируя рост, деление и выживание клеток. Там, где это не нужно, он находится в неактивной форме.

Рисунок 3б. Мутантный K-Ras активен постоянно и приводит к неконтролируемому росту и делению клеток — а это и является раковой опухолью.
Из трех основных белков (K-Ras, N-Ras и H-Ras) K-Ras — наиболее часто мутирующая изоформа [3]. Гены ras мутируют чаще любых других онкогенов и встречаются в 25% опухолей человека: в раке легкого, раке толстой кишки и особенно часто — в раке поджелудочной железы. Это одна из наиболее смертельных опухолей с быстрым метастазированием по организму. За последние 40 лет выживаемость практически не улучшилась: в последние годы в США свой диагноз переживают на пять лет всего лишь 10% пациентов, и всего 3% доживают до более поздних стадий [4]. Мутации K-Ras встречается в более 80% таких опухолей, и, скорее всего, необходимы для развития большинства этих раков. Хотя мутации K-Ras при раке легких и толстой кишки встречаются реже, такие опухоли более агрессивны и хуже поддаются лечению.
Ингибиторы Ras
Несмотря на то, что K-Ras — чаще всего мутирующий онкоген, с момента его открытия в 1982 году прошло более 30 лет до начала клинического исследования первого ингибитора. Одна из главных проблем создания ингибиторов Ras — это присутствие в клетке огромного количества ГТФ (гораздо больше, чем ГДФ), связывающейся с Ras с крайне высоким сродством. Создать ингибитор, который бы вытеснял ГТФ, оказалось крайне сложно, так как смещение равновесия в такой реакции требует молекулы с константой связывания на несколько порядков большей, чем у ГТФ/Ras. Кроме кармана, который всегда связан с нуклеотидфосфатами, белок, казалось, лишен каких-либо мест для связывания ингибитора: в разных статьях K-Ras сравнивали то с бильярдным шаром, то со Звездой Смерти.
Однако в 2013 году вышла статья, в которой группе Кевина Шоката удалось ингибировать K-Ras, найдя еще одно место для связывания [5]. Шокат специализировался на идентификации субстратов ферментов, прежде всего киназ, комбинируя генетическую инженерию и использование химических ингибиторов (область, которую он назвал хемогеномикой). До поиска ингибиторов K-Ras Шокат уже успешно взаимодействовал с фармкомпаниями, разработав ингибиторы PI3K и основав компанию Intellikine, затем проданную Takeda. Для поиска ингибиторов K-Ras лаборатория Шоката воспользовалась методом на основе скрининга фрагментов (fragment based screening), в котором используются молекулы, меньшие по сложности и размеру, чем те, что представлены в химических библиотеках для скрининга. Фрагментоподобные молекулы имеют гораздо меньше активных групп для связывания с белком по сравнению с полноценной лекарственной молекулой, и поэтому сила связывания у них гораздо меньше. Чтобы обнаружить такие взаимодействия, обычно используют очень чувствительные биофизические методы, такие как ЯМР, круговой дихроизм, изотермическую калориметрию или масс-спектрометрию.
Описанные технологии применяются в компьютерном драг-дизайне: «Драг-дизайн: как в современном мире создаются новые лекарства» [6], «Виртуальные тропы реальных лекарств» [7].
Усилия стоят того: полученные фрагменты легче модифицировать до полноценной молекулы, а несколько фрагментов, связывающихся в разных местах одного и того же сайта, можно соединить вместе, получив мощный ингибитор. Одна из мутантных форм K-Ras предоставила исследователям еще одну возможность — в мутанте G12C глицин замещается на гораздо более активный цистеин. Тиольная (или сульфгидрильная) группа цистеина при депротонировании ионизируется, образуя отрицательно заряженную тиолатную группу (рис. 4), которая легко окисляется и связывается с электрофильными (положительно заряженными) молекулами, образуя с ними прочные ковалентные связи [8]. Ковалентные ингибиторы связываются со своими мишенями необратимо, а потому долгое время не пользовались популярностью в медицинской химии. Однако так как мутированный цистеин присутствует только в опухолевых клетках, потенциальный ингибитор будет мало токсичен для клеток с K-Ras дикого типа.

Рисунок 4. Структура цистеина с обозначением тиольной группы и ее превращение в тиолатную группу при депротонировании. Цистеин, возникающий при мутации G12C является главной мишенью ингибиторов K-Ras.
В первичном скрининге с помощью масс-спектрометрии обнаружили два активных фрагмента, один из которых модифицировали, увеличив его активность более чем в четыре раза. Кристаллизация активной молекулы вместе с белком показала, что она связывается с ранее не описанным карманом, который присутствует только в неактивной (ГДФ-связанной) форме K-Ras. Несмотря на то, что мутированные формы Ras в основном находятся в активном ГТФ-связанном состоянии, дальнейшие работы показали, что белок продолжает переключаться между двумя формами, хотя и делает это реже. Сайт связывания назвали switch II pocket. Первые обнаруженные фрагменты связывались с K-Ras обратимо, но в той же статье были найдены электрофильные акриламиды, которые связывались с тем же сайтом необратимо и запирали его в неактивной форме (рис. 5) [5].

Рисунок 5. Первый активный фрагмент, найденный в скрининге (6H05), и ковалентный ингибитор K-Ras (вещество 12). Справа показано связывание 6H05 c K-Ras, а также: мутированный цистеин, карманы Switch I и Switch II.
Несмотря на сравнительно плохую константу связывания, вещество 12 проявляло некоторую активность в клетках и селективно предотвращало рост опухолей с мутацией G12C, но не другими мутациями K-Ras.
В следующих работах той же группе удалось улучшить активность своих ингибиторов и, наконец, они показали достаточную активность в опухолях. Модификации вещества 12 позволили обеспечить плотное связывание с еще одной частью K-Ras — т.н. гидрофобным карманом — и оптимизировать связь с мутированным цистеином. Вещество ARS 853 было уже в 600 раз активнее (рис. 6)! В этой работе наконец удалось полноценно показать все клеточные эффекты ингибитора K-Ras — в клетках с мутацией G12C выключалась активность множества мишеней Ras (таких как Erk, Akt и S6) и прекращалась пролиферация, в то время как в клетках с другими мутациями активность полностью отсутствовала. Однако получившаяся молекула была метаболически нестабильна (время полувыведения меньше 12 минут) и обладала низкой оральной биодоступностью. Решить эту проблему удалось за счет множества модификаций ARS 853, убрав метаболически нестабильные группы и переведя молекулу на хиназолиновый каркас (рис. 7).
 и Amgen")
Рисунок 6. Ингибиторы Шоката (ARS 853) и Amgen на фоне структуры K-Ras с мутированным цистеином, доменом Switch II и так называемым скрытым карманом. См. также рисунок 7.

Рисунок 7. Эволюция ингибиторов K-Ras. Мутированный цистеин намертво связывает акриламидная группа, присоединенная на общий каркас. Особенность AMG510 — группа, взаимодействующая со «скрытым карманом»; у ингибитора Mirati нафтильная группа удобно располагается в гидрофобном кармане. Звездочками отмечены модификации, стабилизирующие MRTX849, — фтор защищает акриламидную группу от глутатион-S-трансферазы, а модификация нафтильной группы — от реакций сульфатации и глюкуронидации.
Спринт до клиники
Работа лаборатории Шоката запустила гонку за новыми ингибиторами K-Ras. Компании Amgen и Mirati Therapeutics пустились вдогонку за основанными Шокатом Araxes Pharma и Wellspring Biosciences и, используя тот же фрагментный скрининг и предыдущие наработки, разработали собственные ингибиторы (см. рис. 7). Araxes продолжила оптимизировать ARS 1620 совместно с Johnson & Johnson, однако их наработки для получения клинического кандидата (и даже его структура) до сих пор не опубликованы.
Группа Amgen начала свои работы по поиску ингибиторов параллельно с первыми работами Шоката и активно использовала его наработки из публикаций и патентной литературы. Главный успех компании заключался в нахождении нового «скрытого» сайта связывания (cryptic pocket), взаимодействие с которым дает еще лучший эффект. Параллельно оптимизируя эти молекулы для повышения активности, фармакокинетических свойств и оральной биодоступности, Amgen получила своего клинического кандидата с кодом AMG510 [9].
Mirati Therapeutics совместно с компанией Array Biopharma также активно использовала наработки Шоката. Их усилия сосредоточились на введении нафтильной группы, улучшавшей связывание в гидрофобном кармане [10]. Дальнейшие усилия компания сосредоточила на оптимизации метаболической стабильности. Для этого они идентифицировали метаболиты, образующиеся при инкубации вещества с мышиными гепатоцитами, и модифицировали уязвимые группы в молекуле. Получившийся клинический кандидат, MRTX849 (рис. 7), обладал высокой стабильностью, сохранив очень высокую активность и специфичность [11].
Всего через шесть лет после первой публикации три ингибитора — AMG 510, MRTX849 и JNJ-74699157 (ARS-3248), — разработанные самим Шокатом, достигли клинических испытаний. Мутация G12C чаще всего встречается в немелкоклеточном раке легкого и раке толстой кишки, и лишь изредка в других опухолях, поэтому все три компании сосредоточились на этих двух болезнях.
Первые данные для AMG 510 и MRTX849 (препараты назвали соторасибом и адаграсибом соответственно) вызвали смешанную реакцию. В исследовании по раку легкого соторасиб прекратил рост опухолей у всех пациентов, а у половины опухоли уменьшились. Однако опухоли кишечника только прекратили рост, практически не сократившись в размерах. Сходные данные в том же году были обнародованы и компанией Mirati. С одной стороны, пациенты в этом испытании имели очень агрессивные опухоли и уже прошли все предыдущие линии химиотерапии — в менее безнадежных случаях, особенно в сочетании с другими препаратами, можно было рассчитывать на лучший эффект. С другой стороны, как обычно бывает в онкологии, вместо ожидаемой революции получился лишь небольшой шаг вперед.
Данные из клинических испытаний продолжали поступать на конференциях. Amgen пришел к финишной прямой первым. В 2021 году FDA условно одобрил соторасиб для терапии немелкоклеточного рака легкого. Данные испытания второй фазы оказались чуть более скромными, чем исходные данные первой фазы: опухоли сократились у 36% участников, и медианная продолжительность ответа на терапию составила около десяти месяцев, однако для таких сложных опухолей и этого оказалось достаточно. Одним из главных преимуществ ингибиторов K-Ras G12C, как и ожидалось, оказалась низкая токсичность: у 11% пациентов отмечались тяжелые побочные эффекты и лишь у 2% пациентов — серьезные побочные эффекты.
Для сравнения, при химиотерапии, например, доцетаксела у таких пациентов может наблюдаться больше 40% тяжелых побочных эффектов, связанных с лечением [12].
Клинические реалии
Итак, первые клинические данные показали, что одного лишь ингибирования Ras недостаточно для полной победы над K-Ras-мутированными опухолями — были обнаружены мутации K-Ras, приводящие к резистентности к ингибиторам; и активация независимых от него сигнальных путей из-за мутаций в других белках. Чтобы добиться полной регрессии опухоли, явно нужны комбинации с другими противоопухолевыми агентами. Для их выбора Amgen обратилась к хорошо исследованной биологии Ras-зависимых сигнальных путей и собственным доклиническим данным. Комбинации с другими ингибиторами Ras-зависимых путей и цитотоксической химиотерапией показали хорошее синергическое действие, однако самым интересным оказалось сочетание ингибиторов K-Ras с иммунотерапией ингибиторами PD1 , активирующими иммунную систему: в такой комбинации AMG510 вызывал полную регрессию опухолей у мышей. По всей видимости, одной из активностей K-Ras является иммуносупрессия, и соторасиб увеличил продукцию цитокинов и инфильтрацию клеток иммунной системы в опухоль [13]. Таким образом, с одной стороны, опухоли лишили способности сопротивляться воздействию иммунной системы, с другой — иммунную систему активировали, «отключив тормоза» в виде PD1/PD-L1 .
PD-1 и PD-L1 относятся к так называемым иммунным контрольным точкам (immune checkpoint). Их повышенная представленность на клетках опухолей и иммунной системы ассоциирована с иммуносупрессией, а их ингибиторы произвели революцию в лечении многих видов рака. В 2018 году за их открытие вручили Нобелевскую премиию [14]. Подробнее про иммунные контрольные точки можно прочесть в статье на «Биомолекуле» «Хороший, плохой, злой, или Как разозлить лимфоциты и уничтожить опухоль» [15].
Еще одной выгодной комбинацией стало сочетание с ингибиторами SHP2-фосфатазы, увеличивающих долю ГДФ-связанных K-Ras — а значит, и число мишеней для G12C-ингибиторов. Основываясь на этих данных, Amgen начал клиническое испытание Codebreak 101, исследующее комбинации соторасиба с четырнадцатью таргетными препаратами и химиотерапевтическими режимами. Mirati также начала свои испытания с комбинациями с обычной химиотерапией, SHP2 ингибиторами и иммунотерапией.
Следующие шаги
Возможность ингибировать Ras возродила интерес к этой области. Несколько компаний, в том числе и Mirati, разрабатывают ингибиторы других мутаций K-Ras, прежде всего G12D — самую частую мутацию при неприступном для терапии раке поджелудочной железы. Ряд ингибиторов основывается на принципе непрямого воздействия на K-Ras: через ингибирование GEF-белков и других активаторов Ras. Один такой класс — упоминавшиеся выше ингибиторы SHP2-фосфатазы, активирующей Ras через дефосфорилирование и увеличивающей связь с его мишенью Raf.
Другой класс непрямых ингибиторов — это ингибиторы взаимодействия Ras с его GEF Sos1. Sos1 необходим для замены ГДФ на ГТФ и активации Ras, и его ингибиторы находятся в клинических испытаниях, в том числе и вместе с K-Ras-ингибитором от Mirati. Количество K-Ras также пытаются снизить с помощью антисмысловых РНК, что показало эффективность в небольшом клиническом испытании. Другие подходы к выключению Ras воздействуют на процессы, необходимые для его связывания с мембраной и не дают им прийти в рабочее положение [16]. Ингибирование шаперона PDE6d, нужного для заякоривания K-Ras в мембране, также не дает ему подобраться к рецепторам, через которые он передает сигнал. Разрабатываются и ингибиторы Ras из класса протеолиз-таргетированных химер, о которых мы писали в другой статье [17].
Надежды вылечить неизлечимое
Ингибиторы K-Ras — яркий пример того, как тщательные исследования структуры в сочетании с прорывами в медицинской химии и подходах к скринингу могут покорить даже самые трудные мишени, ранее ускользавшие от химического ингибирования. Будущее терапий на основе ингибиторов Ras дает надежду на изменения в терапии очень трудных для лечения опухолей, прежде всего смертельного рака поджелудочной железы. Многие исследователи, однако, сходятся на том, что потребуется комбинация ингибиторов Ras с ингибиторами других путей, которые активируются в обход Ras. Также перспективно сочетание с иммунотерапией: она поможет снять торможение иммунной системы, и клетки опухоли будет атакованы как снаружи — собственными клетками организма, — так и изнутри — ингибиторами внутриклеточных сигнальных путей.
Литература
- Adrienne D. Cox, Channing J. Der. (2010). Ras history. Small GTPases. 1, 2-27;
- Jonathan M. L. Ostrem, Kevan M. Shokat. (2016). Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 15, 771-785;
- Natalia Mitin, Kent L. Rossman, Channing J. Der. (2005). Signaling Interplay in Ras Superfamily Function. Current Biology. 15, R563-R574;
- Louis Buscail, Barbara Bournet, Pierre Cordelier. (2020). Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 17, 153-168;
- Jonathan M. Ostrem, Ulf Peters, Martin L. Sos, James A. Wells, Kevan M. Shokat. (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 503, 548-551;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Виртуальные тропы реальных лекарств;
- Leslie B. Poole. (2015). The basics of thiols and cysteines in redox biology and chemistry. Free Radical Biology and Medicine. 80, 148-157;
- Brian A. Lanman, Jennifer R. Allen, John G. Allen, Albert K. Amegadzie, Kate S. Ashton, et. al.. (2020). Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem.. 63, 52-65;
- Jay B. Fell, John P. Fischer, Brian R. Baer, Joshua Ballard, James F. Blake, et. al.. (2018). Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with In Vivo Activity. ACS Med. Chem. Lett.. 9, 1230-1234;
- Jay B. Fell, John P. Fischer, Brian R. Baer, James F. Blake, Karyn Bouhana, et. al.. (2020). Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem.. 63, 6679-6693;
- Pasi A Jänne, Alice T Shaw, José Rodrigues Pereira, Gaëlle Jeannin, Johan Vansteenkiste, et. al.. (2013). Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. The Lancet Oncology. 14, 38-47;
- Jude Canon, Karen Rex, Anne Y. Saiki, Christopher Mohr, Keegan Cooke, et. al.. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 575, 217-223;
- Иммунитет без тормозов: Нобелевская премия за антитела против рака (2018);
- Хороший, плохой, злой, или Как разозлить лимфоциты и уничтожить опухоль;
- Valeria Merz, Marina Gaule, Camilla Zecchetto, Alessandro Cavaliere, Simona Casalino, et. al.. (2021). Targeting KRAS: The Elephant in the Room of Epithelial Cancers. Front. Oncol.. 11;
- Голодные химеры: направленный протеолиз в качестве лекарства;
- Roy S. Herbst, Joseph Schlessinger. (2019). Small molecule combats cancer-causing KRAS protein at last. Nature. 575, 294-295;
- Andrew M. Waters, Channing J. Der. (2018). KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med. 8, a031435;
- Lisa Goebel, Matthias P. Müller, Roger S. Goody, Daniel Rauh. (2020). KRasG12C inhibitors in clinical trials: a short historical perspective. RSC Med. Chem.. 11, 760-770.



Комментарии
0Чтобы оставить комментарий, необходимо
войти