Правдивая история о протеогеномике
13 октября 2023
Правдивая история о протеогеномике
- 752
- 0
- 5
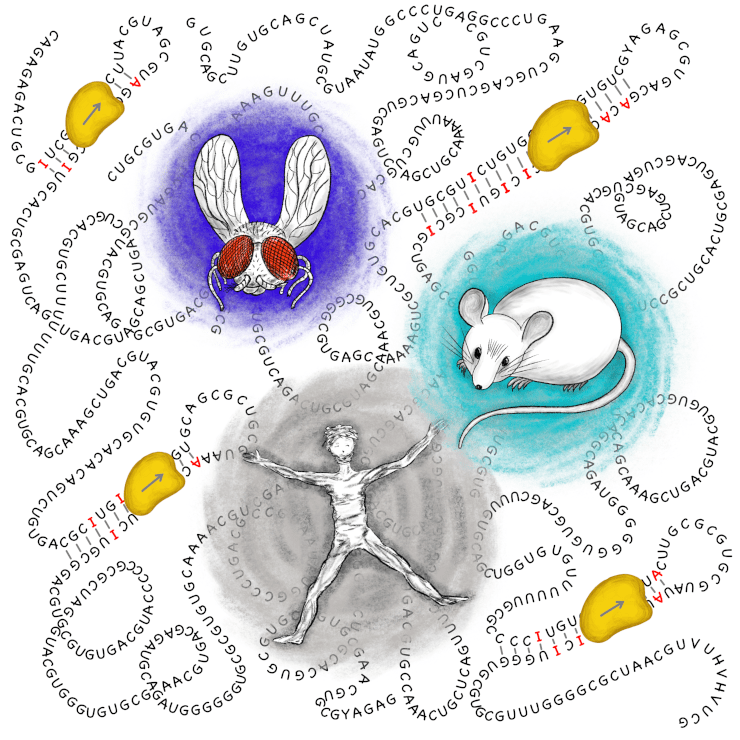
Геном разных людей и даже клеток и тканей в одном организме не одинаковый. Герминальные и соматические мутации вносят изменения, которые зачастую транслируются в белки. Модификации матричных РНК также могут отражаться на последовательностях кодируемых белков. Протеогеномика в широком смысле — это протеомный анализ каждого образца с учетом тех индивидуальных особенностей нуклеиновых кислот, которые могут повлиять на последовательности белков. На иллюстрации показан пример модификации мРНК, кодирующей изменения в белке — редактирование нуклеиновой кислоты путем дезаминирования остатков аденозина (A) с образованием инозина (I).
иллюстрация Ксении Кузнецовой
-
Автор
-
Редакторы
-
Иллюстратор
Узнав о Спецпроекте по мультиомиксным технологиям, который начал выходить на «Биомолекуле», я немного растерялся. Ведь объединение результатов разных омиксных подходов — не особо увлекательная тема. Можно описывать принципы методов, а еще интереснее — новые результаты их использования. Скажем, в современном мире продвижение новых методов как таковых — необходимая с точки зрения коммерческих разработчиков процедура. При этом сами методы бессодержательны с точки зрения фундаментальной биологии или медицины. В музее мы смотрим на статую Венеры Милосской, но мало кто из обычных посетителей интересуется, какими инструментами ее создали. Рационально мыслящие исследователи не станут выбирать способы работы по принципу их новизны или «крутизны». Они возьмут тот резец, которым проще и лучше изваять свою скульптуру, в этом случае — верифицированную или опровергнутую научную гипотезу. Поэтому описание методов как таковых и их сочетаний часто скучны для неспециалистов… Несмотря на все сомнения в полезности мультиомиксов на популярном уровне, сейчас я пишу эти строки. Перефразируя известное высказывание, не сомневается тот, кто ничего не делает. Мы с коллегами почти десять лет назад стали работать в области протеогеномики — интеллектуальной комбинации результатов секвенирования нуклеиновых кислот и панорамного протеомного анализа. Вот поэтому я и считаю неправильным скрывать эту историю от читателей, если могу рассказать о своем опыте из первых рук. Буду писать из головы — выдумывать ничего не придется, как в популярном нынче жанре «тру крайм».

Мультиомиксные технологии
На «Биомолекуле» уже разбирались методы и технологии молекулярной биологии, в том числе протеомные и геномные. В связи с технологическим прогрессом всё чаще применяют их комбинацию, причем к ним добавляются и другие «омики». В спецпроекте «Мультиомиксные технологии» мы расскажем не столько о самих методах, сколько об их применении в самых разных областях — фундаментальной науке, медицине, экологии, сельском хозяйстве. Мы рассмотрим, как калейдоскоп множества методов позволяет лучше увидеть общую картину, ответить на интересующие ученых вопросы и продвинуться в решении практических задач.
Как уже много повторялось в разных материалах «Биомолекулы», омиксы или омики [1], как их слегка неуклюже произносят по-русски, — это производительный анализ разных классов биологических макромолекул. Такой анализ стремятся осуществить в масштабах всей совокупности молекул каждого класса, находящихся в образце (органелле, клетке, ткани, целом организме, если он маленький). Например, липидомика линии раковых клеток в культуре подразумевает как минимум качественный, а лучше количественный анализ максимального технически возможного количества химически различных липидов, содержащихся в клетках этой линии.
Первой омикой была геномика, когда в середине 1990-х годов человечество впервые смогло оперировать не отдельными локусами на хромосомах, а научилось секвенировать целые геномы [2]. Остальные стало принято называть постгеномными технологиями. Но в каком смысле они постгеномные? Просто хронологически или здесь есть что-то большее? В этом смысле омики можно подразделить на две группы. В первую входят технологии анализа непосредственных продуктов геномного «гипертекста», то есть генетического кода — различных РНК и белков. Остальные омики анализируют биомолекулы, лишь косвенно связанные с работой генома, но не закодированные в нем структурно. Это метаболиты в широком смысле, а также рассматриваемые отдельно липиды и полисахариды.
В группе «прямых» постгеномных технологий анализ в подавляющем большинстве случаев осуществляется не de novo, а уже с учетом имеющейся геномной информации. Например, идентификация ДНКовых копий (кДНК) молекул РНК в современной транскриптомике осуществляется на основе уже имеющейся геномной информации. Анализ пептидов и белков посредством масс-спектрометрии, который лежит в основе большинства методов протеомики , также эффективен исключительно по мотивам геномной последовательности кодирующих аминокислотную последовательность участков. В масс-спектрометрии это помогает надежно установить связь между масс-спектром и участком белка (peptide-spectrum match, PSM). Используемые в некоторых случаях методы секвенирования de novo белков и пептидов значительно уступают «геномному» поиску по масс-спектрам в производительности и в покрытии протеома.
Отдельно от масс-спектрометрических методов для идентификации белков в масштабе целого протеома широко используются большие наборы аффинных реагентов, реализованные в формате микрочипов или микропланшетных наборов. Наиболее известные из них — Somalogic на основе аффинных аптамеров (связывающих белки модифицированные олигонуклеотидов) и Olink на основе мультиплексной иммуно-ПЦР.
По техническим причинам белки для масс-спектрометрического анализа, как правило, нужно расщеплять на более короткие пептиды. Подробно этот подход я описываю в материале «12 методов в картинках: Протеомика» [3].
«Косвенные» постгеномные технологии при анализе не опираются на последовательность генома. Как правило, для их эффективного осуществления должно быть известно заведомо, какие метаболиты, липиды или полисахариды могут теоретически содержаться в каждом образце. Например, очень многие метаболиты человека были идентифицированы в эпоху зарождения биохимии в первой половине XX века, когда представления о наследственности были достаточно смутными, а ее материальная основа не была установлена.
Возвращаясь к постгеномным технологиям первой группы, нужно отметить, что транскриптомика и протеомика, почти невозможные без сопровождающей анализ геномной последовательности, мультиомиксны по своей природе. Далее в этом рассказе я сфокусируюсь на протеомике, о которой — так уж вышло — знаю гораздо больше.
Протеогеномика злокачественных опухолей
Итак, аминокислотная последовательность пептидов — продуктов расщепления белков — может быть определена по масс-спектру фрагментов этих пептидов, образованных прямо внутри вакуумной ячейки масс-спектрометра, куда этот пептид попал в ионизованном виде. Получается своего рода мозаика фрагментов с разной молекулярной массой, из которых в итоге складывается последовательность. Интересно, что одними из первых, если не первыми, масс-спектрометрию для секвенирования пептидов использовали советские исследователи под руководством знаменитых Шемякина и Овчинникова более полувека назад [4].
К сожалению, вариантов трактовки масс-спектров фрагментации обычно слишком много, из-за чего надежность образования пары пептид—спектр (peptide-spectrum match) может быть недостаточной. Чтобы уменьшить количество возможных вариантов, рассматривают только те пептиды, которые относятся к белкам, кодируемым в геноме организма, который мы исследуем. К примеру, в практике анализа человеческого протеома используют усредненный, консенсусный геном в некотором выбранном варианте, например, скомпонованный для протеомики на сайте Uniprot или подобном биоинформатическом ресурсе. Однако геномы разных людей, если это не однояйцевые близнецы, не равны. Тем не менее большинство индивидуальных различий находится за пределами кодирующих белки экзонов. Значит, полиморфизм аминокислотной последовательности — явление достаточно редкое, и в большинстве случаев им можно пренебречь (рис. 1).
Если только речь не идет об онкологии.

Рисунок 1. Протеомика с использованием усредненной геномной последовательности — «поиск под фонарем». Если ожидается, что в конкретном протеоме есть существенные отклонения от усредненного протеома, как в злокачественной опухоли, использование стандартного подхода оставит «в темноте» важные аминокислотные замены. Ищущий под фонарем слегка напоминает автора этого материала во время учебы в аспирантуре.
иллюстрация Анастасии Самоукиной
В начале текущего века с развитием секвенирования ДНК исследователям стало ясно, что геномы злокачественных клеток содержат значительные количества соматических мутаций. Возникла концепция генома злокачественной опухоли — особенного, отличного от генома человека, у которого эта опухоль выросла. Именно соматические, в редких случаях, герминальные мутации были определены как ведущий молекулярный механизм канцерогенеза. Есть ли среди них те, что кодируют аминокислотные замены? Обязательно, особенно в гипермутированных опухолях — например, в злокачественных меланомах кожи или различных раках легкого. Какую роль они выполняют? Зачастую обеспечивают перенастройку белков с образованием онкогенов (драйверные мутации), в других случаях возникают как побочный эффект нарушения репарации РНК (пассажирские мутации) и могут мешать опухоли (например, меняя ее иммунный статус). А не надо ли их детектировать на протеомном уровне? Хороший вопрос, которым я задался примерно десять лет назад. Не надо ли вместо консенсусного генома при поиске белков опухолевых образцов — от биопсий до стабильных культур — использовать реальные геномы этих образцов со всеми мутациями?
На момент появления такой крамольной мысли нашлась одна статья 2011 года в специализированном журнале Molecular and Cellular Proteomics, где объединенные раковые мутации, известные на тот момент, добавляли в геномную базу данных человека для поиска протеомов [5]. Вскоре после этого надежно обнаруженные раковые мутации объединили в очень полезную базу знаний COSMIC, из которой вытекает Cancer Census — список около 400 генов, играющих патогенетическую роль в развитии разных опухолей. COSMIC стал своего рода плацдармом для разработки специализированных инструментов поиска мутаций на уровне протеома.
Пока мы с коллегами чесали затылки и думали, что делать, а я подал на эту тему грант в только что созданный Российский научный фонд — тема стала горячей. Использование индивидуально подобранных геномных баз данных для протеомного поиска стало называться уже изобретенным к тому времени термином «протеогеномика» . Американский профессор российского происхождения Алексей Несвижский выпустил в журнале Nature Methods концептуальный обзор, обозначивший проблемы и перспективы протеогеномики [6], а затем и в флагманском Nature было опубликовано первое масштабное исследование, где протеогеномный подход применяли для детальной молекулярной характеристики рака толстой и прямой кишки [7]. Подобные крупные проекты стали реализовывать американские исследователи из недавно образованного Консорциума по анализу протеомики опухолей, выстроенного как продолжение грандиозного проекта Атласа ракового генома. Как уже нередко бывало, я думал в правильном направлении, но с некоторым отставанием.
Правильнее было по порядку получения информации назвать этот подход генопротеомикой, но, кажется, протеогеномика благозвучнее. В итоге получилось как в лесоведении, где сосново-березовым называют лес с преобладанием березы, а не сосны.
Накануне этих знаковых событий мы получили грант и принялись воплощать собственный протеогеномный конвейер. Биоптаты опухолей, а тем более клеточные линии достать мы бы могли, но вот организовать в сжатые сроки геномное секвенирование и глубокий протеомный анализ одних и тех же образцов вряд ли было реально. Небогатый и небыстрый российский исследователь в таком случае обращается к биоинформатике. Открытые данные — настоящее спасение для таких случаев. К тому времени подоспели очень надежные данные секвенирования экзомов 60 раковых клеточных линий из коллекции National Cancer Institute [8]. В другой работе были получены и обнародованы технически продвинутые на тот момент протеомы тех же клеток [9], хотя ее авторы не обращали внимания на мутации, исследуя только количественные характеристик белков без учета аминокислотных замен. Как и в большинстве работ, для протеомного поиска они использовали усредненный геном человека.
На ловца и зверь бежит! Сделаем из экзомов клеточных линий, обнародованных в первой статье, индивидуальные базы данных теоретических белков с мутациями и произведем по ним, а не по усредненному экзому, поиск настоящих белков, фактически присутствующих в протеомах этих линий. Последние в открытом доступе любезно разместили авторы второй статьи.
Казалось бы, что тут сложного — запустили поиск, нашли мутантные пептиды, вот они! Тем не менее, большая проблема в таком поиске — надежность идентификации отдельных пептидов. Как я уже писал раньше в большой статье о протеомике [3], сопоставление теоретических и наблюдаемых масс-спектров для идентификации пептида — это не измерение, а предсказание. У предсказания имеется заданный уровень ложноположительных идентификаций — это обычно 1%. Пептиды составляются в белки, и если в составе белка много идентифицированных пептидов, то ошибки будут нивелироваться. Опаснее ситуация, если белок олицетворяется всего одним пептидом, или вы ищете точечную мутацию, которая тоже будет покрыта одним пептидом. Десять лет назад нужно было доказать, что протеомный подход вообще будет применим для идентификации аминокислотных замен. Скептики предполагали, что правильные результаты могли погрязнуть в ложных идентификациях.
Для верификации вычислительного подхода по идентификации раковых мутаций в протеоме мы применили следующий прием. База данных для поиска содержала аминокислотные замены всех клеточных линий коллекции NCI-60. При поиске в протеоме каждой клеточной линии можно было оценить долю «своих» мутаций, а идентифицированные «чужие» — это как раз ложноположительные результаты. В итоге каждый раз убедительно побеждали правильные варианты (рис. 2), хотя доля неправильных идентификаций многократно превышала расчетный 1% [10]. Эти данные убедительно продемонстрировали эффективность протеогеномного подхода для поиска аминокислотных замен, в частности, раковых мутаций в масштабах протеома, что по тому времени было важным в этой области результатом.

Рисунок 2. Доля найденных мутантных пептидов, относящихся к своему геному, в протеомах клеточных линий коллекции NCI-60. Крупные точки соответствуют «своим» мутациям, мелкие — «чужим». Как видно, свои мутации уверенно побеждают во всех случаях. Это значит, что протеогеномная методика работает. Плохая новость в том, что количество ошибок пока значительно, и истинный уровень ложноположительных результатов равен не расчетному 1%, а примерно 10–15%.
статья с участием автора материала [10] в адаптации Анастасии Самоукиной
Впоследствии мы не раз возвращались к методической части идентификации аминокислотных вариантов в масштабах протеомов. Идентифицируемые не сверхнадежно пептиды с вариантами требовали дополнительного подтверждения. Недавно мы предложили для этого метод протеомного покрытия, где с использованием нескольких протеолитических ферментов одна и та же аминокислотная замена покрывается пептидами различной длины [11]. Уровень ложноположительных идентификаций за счет этого многократно уменьшается. Принцип повышения надежности за счет перекрывающихся неидентичных считываний широко используется при поиске вариантов нуклеотидной последовательности в секвенировании нового поколения, так что в этой работе мы, как могли, пытались адаптировать этот принцип к протеомике (рис. 2).

Рисунок 3. Принцип метода протеомного покрытия для более надежной идентификации аминокислотных вариантов. Для протеомного анализа в большинстве случаев белки расщепляют протеазами на более мелкие фрагменты-пептиды, чтобы повысить чувствительность метода. Классическая протеаза, используемая для этого — трипсин. Если взять три или более протеаз с разной специфичностью, один и тот же участок может быть покрыт пептидами разной длины. Независимые события, подтверждающие одну и ту же аминокислотную замену, существенно увеличивают достоверность ее обнаружения. На рисунке показаны три пептидa якорного белка протеинкиназы А 12 (AKAP12), зарегистрированные путем хромато-масс-спектрометрии в протеоме при его обработке комбинацией трипсина и бактериальной протеазы LysC, одной LysC и еще одной бактериальной протеазой GluC. Так мутация с заменой лизина на глутамин в 216 положении белка (K216Q) в модельных клетках HEK-293 была убедительно идентифицирована методом покрытия протеома.
статья с участием автора материала [11] в адаптации Анастасии Самоукиной
Протеотранскриптомика редактирования РНК
Опередить американцев с их мегапроектами в раковой области нам вряд ли удалось бы, и, научившись идентифицировать аминокислотные замены в протеомах, моя группа стала искать новую нишу в области протеогеномики. В то время мы написали небольшой обзор по протеоформам [12]. Протеоформы — это набор разных белков, образующихся в результате экспрессии одного гена. Это мутантные белки, варианты альтернативного сплайсинга, а также формы, образующиеся в результате посттранскрипционных модификаций матричных РНК и посттрансляционных модификаций белков. После первого раунда рассмотрения статьи один из рецензентов спросил, а почему мы ничего не пишем о протеоформах, образованных за счет редактирования РНК? К своему стыду, я в то время имел очень смутное представление об этом процессе. Что за редактирование, это что-то вроде генетической инженерии или генного нокдауна?
Оказалось, что под редактированием РНК, открытым еще в конце 1980-х годов, понимают природные посттранскрипционные модификации РНК, осуществляемые за счет работы специфических ферментов. Отличительной особенностью таких модификаций оказывается изменение кода РНК. Например, основание аденин модифицируется и преобразуется в гипоксантин, который меняет комплементарность, более охотно соединяясь не с урацилом, а с цитозином. Когда эти модификации приходятся на кодирующую часть матричной РНК, они могут менять смысл кодонов и осуществлять, таким образом, перекодирование последовательности соответствующих белков.
Наиболее распространенный тип редактирования РНК у животных — это как раз упомянутое выше дезаминирование аденозина с образованием нуклеозида инозина (соответствующее азотистое основание называется гипоксантином). Инозин выполняет функции гуанозина, и полученные в результате мРНК могут кодировать измененные белки, например, с заменой глутамина на аргинин, аргинина на глицин и прочих.

Рисунок 4А. ADAR — эволюционно консервативные ферменты, дезаминирующие остатки аденозина в РНК с образованием инозина. Реакция дезаминирования, осуществляемая ферментами ADAR.
иллюстрация Антона Гончарова в адаптации Анастасии Самоукиной
 на аргинин (Арг)")
Рисунок 4Б. ADAR — эволюционно консервативные ферменты, дезаминирующие остатки аденозина в РНК с образованием инозина. Редактирование мРНК субъединицы 2 глутаматного ионотропного рецептора AMPA-типа с заменой глутамина (Глн) на аргинин (Арг). Отмена редактирования этой РНК в результате экспериментов у модельных грызунов или природных мутаций, инактивирующих фермент ADAR2, у человека ведет к продукции закодированного в геноме варианта. Итоговый рецептор-ионный канал лучше пропускает ионы кальция, в результате чего развивается несовместимый с жизнью эпилептический синдром.
иллюстрация Антона Гончарова в адаптации Анастасии Самоукиной
У животных дезаминирование аденозиновых остатков в РНК осуществляют эволюционно консервативные ферменты семейства ADAR (рис. 4a). Они несут черты существенного сходства даже между насекомыми и млекопитающими, что относит их происхождение далеко в докембрий, в начало истории многоклеточности эукариот. Белки ADAR связываются с теми участками РНК, которые формируют двойную спираль (РНК тоже это делают, но не на всем своем протяжении, как ДНК), и в них или вблизи от них преобразуют аденозиновые остатки.
Зачем же все это нужно организмам? Дезаминирование аденозинов в двухцепочечных областях РНК делает их одноцепочечными, ведь комплементарность нарушается. Вместе с тем, повышенная концентрация такой РНК в клетке может затормозить все клеточные процессы — ведь это может указывать на присутствие в ней вирусных геномов. Животные клетки поддерживают внутри себя баланс уровня двухцепочечной РНК, и в регуляции этого процесса задействованы ферменты ADAR, которые ее обезвреживают до определенного порогового уровня, а также работают по системе отрицательной обратной связи при иммунной реакции интерферонов I типа (рис. 5). В соответствии с этим, отключение одной из изоформ этого фермента у человека, а именно ADAR1 (кодируется геном ADAR), врожденными мутациями ведет к различным опасным аутоиммунным проявлениям, включая энцефалит, вызывая орфанное заболевание — одну из форм синдрома Айкарди — Гутьереса. Более подробно о связи ADAR и иммунитета мы пишем в недавно опубликованном обзоре, поэтому я адресую заинтересованных читателей к нему [13].
Несмотря на то, что у насекомых и других первичноротых групп животных нет системы интерферонов, они также защищаются от вирусов с двухцепочечным РНК-геномом — в основном, при помощи РНК-интерференции. Предполагается, что участие в балансировке противовирусного иммунитета у всех животных — первичная функция ADAR. Тогда при чем тут протеом?

Рисунок 5. Участие редактирования РНК ферментами ADAR в неспецифическом иммунном ответе — ингибирующее действие на каскад интерферонов I типа. Воздействие цитоплазматической изоформы фермента ADAR1 на сигнализацию интерферонов I типа путем инактивации двухцепочечной РНК по принципу отрицательной обратной связи. В ядре двухцепочечные РНК претерпевают дезаминирование, катализируемое ферментом ADAR (1). Клетка воспринимает модифицированные транскрипты как безопасные, поскольку их двухцепочечные фрагменты частично расплетаются. Поэтому их появление в цитоплазме не ведет к активации каскада реагирования на избыток дцРНК (2). Если концентрация дцРНК в цитоплазме повышается в результате вирусной атаки или гиперпродукции собственной дцРНК, например, путем экспрессии Alu-повторов, насыщенных такой конформацией, активируются соответствующие сенсоры дцРНК — например, белки MDA5 и RIG-1 (3). Активация этих сенсоров запускает противовирусный неспецифический иммунитет посредством факторов транскрипции, инициирующих экспрессию интерферонов I типа (4). Аутокринная и паракринная интерфероновая сигнализация ведет к активации экспрессии специального набора интерферон-стимулируемых генов за счет связывания фосфорилированных комплексов факторов транскрипции STAT1/STAT2 с особыми геномными ISRE-элементами (5). В гене фермента ADAR1 интерфероновая сигнализация включает экспрессию изоформы p150, которая, в отличие от ядерной изоформы р110, используемой по умолчанию, мигрирует в цитоплазму и редактирует избыток дцРНК. Это снижает стимуляцию интерферонового каскада по принципу отрицательной обратной связи (6).
обзор с участием автора материала [13] в адаптации Анастасии Самоукиной
Иногда двухцепочечные структуры попадались и в кодирующих белок частях матричных РНК. Занимаясь своей работой, ферменты ADAR разрушали эти структуры и попутно… меняли содержание кодонов. В итоге образовывались перекодированные белки. Большинство из них, вероятно, теряло конформационную стабильность и разрушалось, часть просто сохраняла свою функцию, но в ряде случаев точечные замены вели к образованию протеоформы с новыми, иногда полезными свойствами. Поэтому перекодирующее действие ADAR закреплялось в процессе эволюции. У нас с вами иммунная и перекодирующая функции фермента даже разделены на два разных паралогичных гена — ADAR и ADARB1. Для первого характерна работа в более протяженных двухцепочечных участках РНК, второй же тяготеет к матричным РНК.
Одна из первых работ по подробному разбору перекодирующей функции ADAR касалась головоногих моллюсков. В авторитетном журнале Cell израильские ученые сообщили о невероятной пластичности транскриптома и протеома двух видов осьминогов, кальмара и каракатицы за счет редактирования РНК ферментами ADAR [14]. Позднее в исследовании калифорнийского двупятнистого осьминога (Octopus bimaculoides) было показано, что массированное перекодирование обеспечивает адаптацию нервной системы этого головоногого к существенным изменениям температуры воды, которые он способен переносить [15]. Шутка ли, активность ADAR наблюдали в 13 тысячах кодонов, во многих из которых происходили аминокислотные замены! Молекулярная эволюция головоногих моллюсков в свете столь масштабного редактирования их транскриптома после появления первых работ в этой области находится в фокусе новых исследований, к которым, например, присоединились биоинформатики из лаборатории М.С. Гельфанда [16].
Существенные изменения, как было показано в 2010-е годы, вносятся редактированием и в транскриптом плодовой мушки. Мы впервые получили протеомы головного мозга этого модельного насекомого — за неимением свежих каракатиц — и обнаружили в них на белковом уровне примерно 70 сайтов перекодирования из 1300 теоретически возможных [17]. Изменениям подвергался, в частности, аппарат высвобождения синаптических везикул в постсинаптической мембране, по аналогии с подобными же изменениями у головоногих моллюсков (рис. 6). По аналогии стоит предполагать, что пойкилотермные насекомые, способные размножаться в интервале почти 20 °C, при помощи перекодирования белков пресинаптической мембраны осуществляют термальную адаптацию. Кроме того, интенсивные изменения в редактировании транскриптома и соответствующем перекодировании протеома сопровождали метаморфозы дрозофилы в течение ее жизненного цикла [18].

Рисунок 6. SNARE-комплекс, обеспечивающий слияние синаптических пузырьков с пресинаптической мембраной при активации синапса потенциалом действия. У дрозофилы как минимум три белка этого комплекса подлежат перекодированию за счет редактирования РНК. Как предполагается, так обеспечивается адаптация нервной системы насекомого к изменяющейся температуре окружающей среды.
Wikipedia в адаптации Анастасии Самоукиной
Мушка-дрозофила — прекрасный, горячо любимый нами объект исследования [19], но мы всегда работали в институтах со словом «медицина» в названии. Значит, дальше нужно было искать перекодированные участки в протеомах млекопитающих. Как я писал до этого, функции регуляции уровня эндогенных двухцепочечных РНК у человека возложена на первую изоформу ADAR, а вот перекодированием белков путем изменений в мРНК занимается ADAR2. Мутации в гене последнего, как правило, вообще не совместимы с жизнью, а почему? Оказывается, на модели мыши, что отмена редактирования всего одного кодона ведет к таким тяжелым последствиям. Речь о замене глутамина на аргинин в 607 положении субъединицы 2 глутаматного ионотропного рецептора AMPA-типа (кодируется геном GRIA2). Наличие перекодированного сайта замедляет ток кальция в ионном канале, образуемого этим рецептором. Происходящее еще в эмбриогенезе почти полное перекодирование таких субъединиц обеспечивает нормальное развитие нервной системы, а его отмена ведет к эксайтотоксичности и ранней внутриутробной гибели (рис. 4Б). Описаны и другие единичные случаи перекодирования, имеющие функциональное значение, — например, в филамине А (FILA) или в коатомере А (COPA), но не настолько радикально воздействующие на организм.
Для описания ландшафта перекодирования на уровне целых протеомов мы обработали 40 наборов протеомных данных, относящихся к различным клеткам и тканям человека [20]. Всего использованные данные включали в себя около восьми тысяч индивидуальных запусков хромато-масс-спектрометра. Они оказались действительно большими по объему. Тем не менее, такой набор не совсем отвечал классическому определению Big Data, поскольку его составляющие не были получены в одном и том же формате, как например, траектории полета птиц, поездки на такси или данные кровяного давления у пациентов. Этот недостаток стандартизации — наличие разных устройств и способов, обусловленных выбором исследователей и конкурирующими решениями на рынке — сильно мешает масштабной переобработке и переоценке открытых данных в протеомике. Однако заставить исследователей действовать в едином порыве, как вы понимаете, достаточно сложно.
В отличие от насекомых и моллюсков, число обнаруженных участков перекодирования у человека совсем скромное — не более 20 сайтов, идентифицированных как минимум в двух наборах данных. Среди них можно выделить участки, специфичные для нейрональных белков, и те, которые встречаются по всему организму — вероятно, в кровеносных сосудах, которые, как и нервная ткань, представляют собой «горячую точку» редактирования РНК. Немногие из них охарактеризованы функционально, значение остальных остается загадкой. Исследовать их непросто — необходимы модели, где принудительно внесена соответствующая замена уже на уровне ДНК, иными словами, точечный knock-in, или, наоборот, запрещено редактирование РНК в одном конкретном кодоне, что требует сходных манипуляций на уровне генома.
Вместе с другими исследователями, среди которых нужно выделить большую группу из того же ракового консорциума CPTAC, изучивших перекодирование белков в образцах злокачественных опухолей [21] при помощи протеогеномики (а точнее, протеотранскриптомики), мы насколько возможно исчерпывающе охарактеризовали ландшафт перекодирования белков за счет редактирования мРНК ферментами ADAR у человека.
Неоантигены и противораковые вакцины
Протеогеномный подход, позволяющий более точно охарактеризовать особенности индивидуальных протеомов, учесть кодирующие аминокислотные соматические мутации в геноме и посттранскрипционные модификации в протеоме, представляет собой прекрасный инструмент для фундаментальных исследований. Тем не менее, читатели, а также финансирующие организации, конечно же, вправе задать вопрос, а какое же, позвольте, народнохозяйственное значение имеют ваши штудии?
Даже точечные раковые мутации на белковом уровне могут быть целью для использования таргетной терапии. Примером служит препарат вемурафениб, который действует в тех случаях злокачественой меланомы кожи, когда онкогенная протеинкиназа B-Raf в опухоли содержит аминокислотную замену валина на глутаминовую кислоту в 600-м положении. Конечно, определить уровень мутантного белка посредством масс-спектрометрии было бы в этом случае привлекательно, но в случае хорошо изученных продуктов основанный на ПЦР в реальном времени тест на экспрессию мутантной мРНК, честно говоря, справляется достаточно хорошо для его практического применения и без всяких белков.
Привлекательное и, возможно, прикладное направление использования масс-спектрометрии — это анализ неоантигенов в опухолевых протеомах. Как известно, в процессе развития плода развивающиеся Т-лимфоциты проходят отбор на толерантность к собственным антигенам организма. Клоны с Т-клеточными рецепторами, реакционноспособными в отношении белков собственного тела, подвергаются управляемой клеточной гибели. Однако, как уже упоминалось выше, протеом клеток злокачественной опухоли может изобиловать соматическими мутациями, возникшими во время развития опухоли. Если фрагменты мутантных белков способны представляться клеточному иммунитету в молекулах гистосовместимости, такие белки будут распознаваться как чужеродные, новые антигены. Поэтому такие участки раковых белков называются неоантигенами, а конкретные связывающие Т-клеточные рецепторы последовательности — неоэпитопами. Поиск и идентификация неоэпитопов на белковом уровне — нетривиальная, но вместе с тем и потенциально полезная для медицины задача. По числу неоантигенов в конкретной опухоли можно предсказывать эффективность применения иммунотерапевтических препаратов — ингибиторов контрольных точек . Более того, идентифицированные неоантигены могут непосредственно использоваться для создания персонализированных противоопухолевых вакцин.
Подробнее об этой терапии можно прочитать в материалах «Биомолекулы» [22] и даже посмотреть видео на эту тему [23]. — Ред.
Проведены клинические исследования, в которых пептидные аналоги неоантигенов использовали непосредственно для инъекций, пытаясь стимулировать Т-лимфоциты внутри организма. Например, совсем недавно вакцина на основе пептида, включающего последовательность мутантного гистона H3 с заменой лизина на метионин в 27 положении, показала неплохие результаты в лечении агрессивной опухоли мозга — диффузной глиомы [24], содержащей эту мутацию. Один из пациентов в этом исследовании полностью исцелился и уже около трех лет не проявлял признаков заболевания.
В более изощренном варианте, собственные Т-лимфоциты извлекают из крови пациента и стимулируют их неоантигенами, после чего вводят обратно уже готовыми к интенсивной борьбе с опухолями.
Важно отметить, что набор неоантигенов для каждой опухоли индивидуален, а клинические исследования в этой области направлены на создание персонализированной терапии. В этих случаях изъятую у пациента биопсию исследуют путем секвенирования и хромато-масс-спектрометрии для получения мультиомиксного ландшафта. Неоантигены могут быть предсказаны из транскриптома, из протеома и из так называемого «лигандома», когда масс-спектрометрию используют для анализа пептидов-эпитопов, аффинно связанных с молекулами гистосовместимости. Последний метод наиболее близок к реальному набору неоантигенов, существующих в образце опухоли, но при этом он более трудоемок и требователен к реагентике и количеству материала для анализа. В идеальной ситуации лаборатория оперативно подключается к ведению пациента и вместе с врачами-онкологами выбирает индивидуальную стратегию лечения на основе молекулярного ландшафта опухоли. Сейчас подобные экспериментальные схемы реализованы в некоторых странах в формате клинических исследований I фазы [25], [26].
Любопытно, что два направления протеогеномики, о которых я здесь пишу, соединились в недавно опубликованном исследовании [27]. Оказывается, посттранскрипционные модификации мРНК, включая редактирование ферментами ADAR, также способны приводить к образованию неоантигенов в злокачественных опухолях. Такие случаи были продемонстрированы мультиомиксным методом на трех десятках пациентов с различными типами злокачественных опухолей.
Повторяя сказанное в начале этого материала, признаюсь, что я не сторонник введения новых понятий в науке и технике, особенно там, где без них можно обойтись. Концептуально каждая омика — набор независимо развивавшихся методов с собственным инструментарием. Тем не менее, системное объединение и совместная трактовка их результатов — захватывающая, нередко сложная биоинформатическая задача. В этой статье я старался поделиться с читателем своим скромным опытом трактовки объединенных результатов секвенирования нуклеиновых кислот и хромато-масс-спектрометрии белков в различных модельных объектах и на клиническом материале. Напоследок важно сказать, что ничего из рассказанного мной о собственной работе не могло бы сбыться без моих дорогих коллег — соавторов цитируемых здесь статей, которых я искренне благодарю.
Описанные здесь собственные исследования проведены в 2012–22 годах в моей московской лаборатории вначале в Институте биомедицинской химии им. В.Н. Ореховича, а потом в Центре физико-химической медицины им. Ю.М. Лопухина при деятельном участии коллег из лаборатории Михаила Горшкова в Институте энергетических проблем химической физики им. В.Л. Тальрозе. Работы финансировались грантами Российского научного фонда и Российского фонда фундаментальных исследований.
Литература
- «Омики» — эпоха большой биологии;
- Геном человека: как это было и как это будет;
- 12 методов в картинках: протеомика;
- M. M. SHEMYAKIN, YU. A. OVCHINNIKOV, A. A. KIRYUSHKIN, E. I. VINOGRADOVA, A. I. MIROSHNIKOV, et. al.. (1966). Mass Spectrometric Determination of the Amino-Acid Sequence of Peptides. Nature. 211, 361-366;
- Jing Li, Zengliu Su, Ze-Qiang Ma, Robbert J.C. Slebos, Patrick Halvey, et. al.. (2011). A Bioinformatics Workflow for Variant Peptide Detection in Shotgun Proteomics. Molecular & Cellular Proteomics. 10, M110.006536;
- Alexey I Nesvizhskii. (2014). Proteogenomics: concepts, applications and computational strategies. Nat Methods. 11, 1114-1125;
- Bing Zhang, Jing Wang, Xiaojing Wang, Jing Zhu, Qi Liu, et. al.. (2014). Proteogenomic characterization of human colon and rectal cancer. Nature. 513, 382-387;
- Ogan D. Abaan, Eric C. Polley, Sean R. Davis, Yuelin J. Zhu, Sven Bilke, et. al.. (2013). The Exomes of the NCI-60 Panel: A Genomic Resource for Cancer Biology and Systems Pharmacology. Cancer Research. 73, 4372-4382;
- Amin Moghaddas Gholami, Hannes Hahne, Zhixiang Wu, Florian Johann Auer, Chen Meng, et. al.. (2013). Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Reports. 4, 609-620;
- Maria A. Karpova, Dmitry S. Karpov, Mark V. Ivanov, Mikhail A. Pyatnitskiy, Alexey L. Chernobrovkin, et. al.. (2014). Exome-Driven Characterization of the Cancer Cell Lines at the Proteome Level: The NCI-60 Case Study. J. Proteome Res.. 13, 5551-5560;
- Lev I. Levitsky, Ksenia G. Kuznetsova, Anna A. Kliuchnikova, Irina Y. Ilina, Anton O. Goncharov, et. al.. (2022). Validating Amino Acid Variants in Proteogenomics Using Sequence Coverage by Multiple Reads. J. Proteome Res.. 21, 1438-1448;
- Andrey Lisitsa, Sergei Moshkovskii, Aleksey Chernobrovkin, Elena Ponomarenko, Alexander Archakov. (2014). Profiling proteoforms: promising follow-up of proteomics for biomarker discovery. Expert Review of Proteomics. 11, 121-129;
- Anton O. Goncharov, Victoria O. Shender, Ksenia G. Kuznetsova, Anna A. Kliuchnikova, Sergei A. Moshkovskii. (2022). Interplay between A-to-I Editing and Splicing of RNA: A Potential Point of Application for Cancer Therapy. IJMS. 23, 5240;
- Noa Liscovitch-Brauer, Shahar Alon, Hagit T. Porath, Boaz Elstein, Ron Unger, et. al.. (2017). Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell. 169, 191-202.e11;
- Matthew A. Birk, Noa Liscovitch-Brauer, Matthew J. Dominguez, Sean McNeme, Yang Yue, et. al.. (2023). Temperature-dependent RNA editing in octopus extensively recodes the neural proteome. Cell. 186, 2544-2555.e13;
- Mikhail A. Moldovan, Zoe S. Chervontseva, Daria S. Nogina, Mikhail S. Gelfand. (2022). A hierarchy in clusters of cephalopod mRNA editing sites. Sci Rep. 12;
- Ksenia G. Kuznetsova, Anna A. Kliuchnikova, Irina U. Ilina, Alexey L. Chernobrovkin, Svetlana E. Novikova, et. al.. (2018). Proteogenomics of Adenosine-to-Inosine RNA Editing in the Fruit Fly. J. Proteome Res.. 17, 3889-3903;
- Anna A. Kliuchnikova, Anton O. Goncharov, Lev I. Levitsky, Mikhail A. Pyatnitskiy, Svetlana E. Novikova, et. al.. (2020). Proteome-Wide Analysis of ADAR-Mediated Messenger RNA Editing during Fruit Fly Ontogeny. J. Proteome Res.. 19, 4046-4060;
- Модельные организмы: дрозофила;
- Lev I. Levitsky, Mark V. Ivanov, Anton O. Goncharov, Anna A. Kliuchnikova, Julia A. Bubis, et. al.. (2023). Massive Proteogenomic Reanalysis of Publicly Available Proteomic Datasets of Human Tissues in Search for Protein Recoding via Adenosine-to-Inosine RNA Editing. J. Proteome Res.. 22, 1695-1711;
- Xinxin Peng, Xiaoyan Xu, Yumeng Wang, David H. Hawke, Shuangxing Yu, et. al.. (2018). A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell. 33, 817-828.e7;
- Иммунитет без тормозов: Нобелевская премия за антитела против рака (2018);
- Как развивалась иммунотерапия рака?;
- Niklas Grassl, Isabel Poschke, Katharina Lindner, Lukas Bunse, Iris Mildenberger, et. al.. (2023). A H3K27M-targeted vaccine in adults with diffuse midline glioma. Nat Med;
- Rui Chen, Kelly M. Fulton, Susan M. Twine, Jianjun Li. (2021). IDENTIFICATION OF MHC PEPTIDES USING MASS SPECTROMETRY FOR NEOANTIGEN DISCOVERY AND CANCER VACCINE DEVELOPMENT. Mass Spectrometry Reviews. 40, 110-125;
- Na Xie, Guobo Shen, Wei Gao, Zhao Huang, Canhua Huang, Li Fu. (2023). Neoantigens: promising targets for cancer therapy. Sig Transduct Target Ther. 8;
- Celina Tretter, Niklas de Andrade Krätzig, Matteo Pecoraro, Sebastian Lange, Philipp Seifert, et. al.. (2023). Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification. Nat Commun. 14.



Комментарии
0Чтобы оставить комментарий, необходимо
войти