Прионы: исследования таинственных молекул продолжаются
03 ноября 2012
Прионы: исследования таинственных молекул продолжаются
- 24277
- 0
- 14

Путь прионов
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: Прионные заболевания — феномен, открытый в двадцатом веке, и в нем же начавший играть большую роль: увеличение продолжительности жизни в развитых странах привело к тому, что все больше людей стало доживать до «своего Альцгеймера» или «своего Паркинсона». Природа нейродегенеративных заболеваний продолжает оставаться туманной, и ученые пока исследуют только отдельные их аспекты — например, причину развития именно в старческом возрасте или способность передаваться от одних видов живых существ другим.

«Био/мол/текст»-2012
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2012 в номинации «Лучшее новостное сообщение».
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Всё началось с того, что в 20 веке учёные заинтересовались природой необычных заболеваний человека и животных: куру, Крейтцфельда-Якоба, скрэпи. Заметное сходство патологии этих болезней дало основание для гипотезы об их инфекционности, что впоследствии было экспериментально подтверждено. Тогда возник вопрос о возбудителе данных заболеваний. Прежде чем был найден ответ, были выявлены необычайные свойства возбудителей: они не размножаются на искусственных питательных средах, устойчивы к высокой температуре, формальдегиду, различным видам излучений, действию нуклеаз. Очистка инфекционного материала и его изучение позволило провозгласить о том, что «во всём виноват» белок, который 30 лет назад получил название прион (от англ. pr[otenacious infect]ion — белковая инфекция).
Так, известные американские учёные — вирусолог и врач Д.К. Гайдушек, раскрывший инфекционную природу прионных болезней, в 1976 г. и биохимик С.Б. Прузинер, который определил прионы и разработал прионную теорию, в 1997 г., — были удостоены Нобелевских премий. Их работы стали импульсом для последующих исследований, благодаря которым были изучены новые виды прионных инфекций. Но, даже несмотря на неугасающий интерес к «прионной теме», образование прионов до сегодняшнего дня остаётся загадкой.
Биологическая сущность прионов
Рисунок 1. Метафора нейродегенеративного поражения мозга — это губка, в которую превращается нервная ткань в результате массовой гибели нейронов.
Молекула приона не является чем-то экзотическим: в «нормальной» форме она имеется на поверхности нервных у каждого из нас. При этом мы отлично себя чувствуем, и наши нервные клетки живы и здоровы. Однако это всё до тех пор, пока наш нормальный белок не «переродится» в аномальную форму. А если это случится, то приведёт к ужасающим последствиям: инфекционная форма прионов имеет свойство «склеиваться» с другими молекулами и, мало того, «конвертировать» их в эту же самую форму, вызывая «молекулярную эпидемию». В результате этой полимеризации на нервной клетке появляются токсичные белковые бляшки , и она погибает [1]. На месте погибшей клетки образуется пустота — вакуоль, заполненная жидкостью. С течением времени будет исчезать один нейрон за другим, а в мозге — образовываться всё больше «дыр», пока, наконец, мозг не превратится в губку (рис. 1), за чем неминуемо последует смерть.
Существует упрощенное представление, что полимеризованные прионные фибриллы «протыкают» нейрон, что вызывает его гибель. На самом деле это не совсем так: предшествующие фибриллярной стадии сферические агрегаты прионов также обладают токсичностью (по крайней мере, для болезни Альцгеймера): «Альцгеймеровский нейротоксин: ядовиты не только фибриллы». — Ред.
Но как может нормальный природный белок (обозначается PrPC) вдруг стать патологическим (PrPSc; Sc — от слова «scrapie»)? Что должно произойти? Как и в случае «обычной» инфекции, для такой трансформации необходима встреча с молекулой инфекционного приона. Существуют два пути передачи этой молекулы: наследственный (за счёт мутаций в гене, кодирующего белок) и инфекционный. То есть внедрение приона может произойти неожиданно — например, при употреблении в пищу недостаточно хорошо прожаренного или сваренного мяса (в котором должна присутствовать форма PrPSc), при переливании крови, при трансплантации органов и тканей, при введении гормонов гипофиза животного происхождения.
И тогда происходит удивительное событие: нормальные молекулы белка, контактируя с патологическими, сами превращаются в них, изменяя свою пространственную структуру (механизм трансформации остаётся загадкой и по сей день) [1]. Таким образом прион, как самый настоящий инфекционный агент, заражает нормальные молекулы, запуская цепную реакцию, разрушительную для клетки.
Некоторые сведения о прионах
Исследователи отмечают:
- прионный белок включает 254 аминокислотных остатка и «весит» 33–35 килодальтон [2];
- ген, кодирующий белок PrP, найден у человека, млекопитающих и птиц [1];
- для полного уничтожения прионного белка нужна температура не менее 1000 градусов [1]!
- возможно, прионы принимают участие в межклеточном узнавании и клеточной активации [3];
- не исключено, что функцией прионов является подавление возрастных процессов [3];
- при развитии клинических проявлений прионных заболеваний нет ни признаков воспаления, ни изменений в крови;
- предполагается, что прионы участвуют в развитии шизофрении и миопатии;
- механизм действия прионов и их превращения из нормальной формы в патологическую остаётся неясным.
Условия возникновения заболеваний
Условия возникновения прионовых болезней уникальны. Они могут формироваться по трём сценариям: как инфекционные, спорадические и наследственные поражения. В последнем варианте главную роль играет генетическая предрасположенность [2].
Знаменитый исследователь прионов Стэнли Прузинер (Stanley Prusiner) выделяет две поразительные особенности, присущие таким нейродегенеративным заболеваниям, как болезнь Крейтцфельда-Якоба, болезнь Альцгеймера и болезнь Паркинсона. Первая заключается в том, что более 80% случаев заболевания — спорадические (то есть, случайные, возникающие «сами собой»). Вторая: несмотря на то, что большое количество мутантных белков, специфичных к определённой болезни, экспрессируется в процессе зародышевого развития, формы наследования этих нейродегенеративных заболеваний проявляются позже. Это предполагает, что некоторые процессы происходят во время старения, которое «дает волю» болезнетворным белкам [5]. Более 20 лет назад автор утверждал, что данный процесс включает случайный рефолдинг (пересворачивание) белка в неправильно свёрнутый, что соответствует переходу в инфекционное состояние — прион.
Интересные факты насчет болезни Альцгеймера: ее вероятность может повышаться вследствие хронического недосыпания («Новый шаг к пониманию болезни Альцгеймера: возможно, недосыпание является одним из факторов риска»), а сам альцгеймеровский нейропептид (β-амилоид Aβ) может быть частью системы врожденного иммунитета («Возможно, β-амилоид болезни Альцгеймера — часть врождённого иммунитета»). — Ред.
В последнее десятилетие интерес к этой теме возобновился в связи с возможностью развития диагностики и эффективной терапии [5]. Появилось множество различных объяснений для возрастных нейродегенеративных болезней, — например, окислительная модификация ДНК, липидов и/или белков; соматические мутации; измененный врождённый иммунитет; экзогенные токсины; несоответствия ДНК—РНК; нарушение работы шаперонов; отсутствие одного из аллелей гена [5]. Альтернативным комплексным разъяснением служит то, что различные группы белков могут формировать прионы. Несмотря на то, что небольшое количество прионов может быть удалено посредством путей белковой деградации, их чрезмерное накопление с течением времени позволяет прионам самостоятельно распространяться в организме (рис. 2), что приводит к нарушению деятельности центральной нервной системы [5].

Рисунок 2. Процессы нейродегенерации, вызванной прионами. Сверху: накопление «нормального» прионного белка повышает его вероятность перехода в токсичную конформацию, которая описывается бóльшим содержанием β-структуры. Прионы наиболее патогенны в форме олигомеров; после образования фибрилл токсичность снижается. В зависимости от того, о каком конкретно прионном белке идет речь, в патологическом состоянии он может образовывать бляшки, клубки или тельца включения. Возможные пути лекарственного вмешательства: (I) снижение концентрации «нормального» белка-предшественника; (II) ингибирование образования прионной формы; (III) уничтожение токсичных агрегатов. Снизу: Наследственная старческая нейродегенерация объясняется двумя событиями: наличием мутантной формы предшественника и образованием из него приона, готового к олиго- и полимеризации с образованием токсичных форм.
Группы риска прионных заболеваний
Вот кого прионные заболевания могут настичь с наибольшей вероятностью:
- работники пищевой промышленности;
- ветеринары;
- патологоанатомы;
- хирурги;
- пациенты трансплантолога;
- каннибалы;
- лица, в семье которых были замечены синдромы Герстманна—Штрейслера—Шейнклера или фатальной инсомнии.
Лабораторная диагностика и лечение
Диагностика базируется на внутримозговом заражении мышат или хомяков, у которых медленно (до 150 дней) развивается соответствующее заболевание, если пациент был болен [2]. Часто проводится гистологическое исследование головного мозга погибших животных [2].
К сожалению, до настоящего времени еще не разработаны эффективные методы лечения прионовых болезней, хотя попытки предотвратить конформационный переход нормального белка в аномальный производятся. Поэтому самым надёжным способом предупреждения развития инфекционных форм является профилактика [2].
Особенно актуальным становится решение «прионного вопроса» в связи с нарастающей угрозой возникновения эпидемии через инвазивные медицинские операции и даже при приёме лекарственных средств.
Перспективы
По-видимому, интерес к прионам не угаснет до тех пор, пока предположения на их счёт полностью не подтвердятся и не будут найдены эффективные способы лечения прионных заболеваний. В статье [6] говорится о необходимости современного исследования, которое требует тщательного рассмотрения чужеродных прионов в экстраневрональных тканях.
В качестве модельных объектов авторы использовали мышей: две линии, которые трансгенно экспрессировали овечий прионный белок, и одну линию, которая экспрессировала человеческий прионный белок (рис. 3). Задачей было сравнить эффективность межвидовой передачи инфекции посредством тканей мозга и селезёнки. Внутримозговое заражение чужеродным прионным белком выражалось в отсутствии или небольшом количестве инфекционного агента в мозгах этих мышей. Однако инфекционные чужеродные прионы обнаруживались в селезёнке на более ранних этапах заражения в сравнении с моментом, когда были использованы нейротропные прионы, тем самым определяя, что лимфатическая ткань может быть более пермиссивной к распространению чужеродных прионов по сравнению с мозгом.
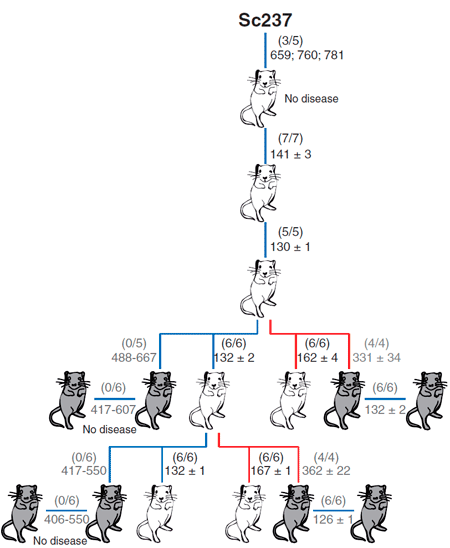
Рисунок 3. Способность приона хомяков Sc237 заражать и передаваться при введении в мозг или селезенку трансгенным мышам, имеющим прионный белок PrP овцы (tg338; белые мыши) или человека (tg7; серые мыши). Число заболевших/инъецированных мышей показано в скобках; ниже приведено среднее время жизни (в днях).
Чем вызвана эта предпочтительная репликация прионов в лимфатических тканях, пока неизвестно. Однако полученные данные показывают, что человек может быть более чувствительным к чужеродным прионам, чем предполагалось ранее на основании присутствия прионов в мозгу, и по этой причине бессимптомный переносчик прионной болезни может быть не распознан. Это ещё раз подтверждает, что такая могущественная биомолекула как прион таит в себе немало загадок, раскрытие которых, возможно, поможет в понимании ряда неразрешимых проблем человечества...
Литература
- Абрамова З.И. Исследование белков и нуклеиновых кислот. Казань: Казанский государственный университет, 2006. — 157 с.;
- Новиков Д.К., Генералов И.И., Данющенкова Н.М. Медицинская микробиология. Витебск: ВГУ, 2010. — 597 с.;
- Прудникова С.В. Микробиология с основами вирусологии. Красноярск: ИПК СФУ, 2008;
- Поздеев О.К., Покровский В.И. Медицинская микробиология. М.: Гэотар-мед, 2001. — 765 с.;
- S. B. Prusiner. (2012). A Unifying Role for Prions in Neurodegenerative Diseases. Science. 336, 1511-1513;
- V. Beringue, L. Herzog, E. Jaumain, F. Reine, P. Sibille, et. al.. (2012). Facilitated Cross-Species Transmission of Prions in Extraneural Tissue. Science. 335, 472-475;
- Carolina Pola. (2012). Prion escape to spleen. Nat Med. 18, 360-360;
- Элементы: «10 фактов о прионах и амилоидах»;
- Элементы: «Геометрия белковых тел»;
- Charles Weissmann. (2012). Mutation and Selection of Prions. PLoS Pathog. 8, e1002582.



Комментарии
0Чтобы оставить комментарий, необходимо
войти