Неуловимая архитектура хроматина мухи
04 января 2021
Неуловимая архитектура хроматина мухи
- 762
- 0
- 5
Архитектура хроматина одиночных клеток плодовой мушки дрозофилы оказалась намного более предсказуемой, чем ожидалось
-
Автор
-
Редакторы
-
Рецензенты
Статья на конкурс «Био/Мол/Текст»: Судьба клетки во многом определяется тем, как ее гены, закодированные в ДНК, считываются и работают. Мы узнаём всё больше о том, как в этом процессе важна пространственная организация хроматина (иными словами — его архитектура). Структурная биология хроматина — большая и кипучая область, и именно здесь можно придумать и поставить такой эксперимент, который прояснит фундаментальные принципы жизни клетки. Мне удалось стать не только свидетелем, но и участником такого события. С помощью непростого эксперимента фиксации конформации хромосом одиночных клеток мы предположили два возможных механизма формирования структуры хроматина мухи, тесно связанных с активностью генов. Более того, мы выяснили ряд особенностей хроматина этого организма. На пути к открытию пришлось преодолеть всевозможные трудности. И теперь, когда работа опубликована в Nature Communications, я делюсь рассказом о том, почему это было не только трудно, но и интересно.

Конкурс «Био/Мол/Текст»-2020/2021
Эта работа опубликована в номинации «Своя работа» конкурса «Био/Мол/Текст»-2020/2021.

Партнер номинации — Российский научный фонд.
Генеральный партнер конкурса — ежегодная биотехнологическая конференция BiotechClub, организованная международной инновационной биотехнологической компанией BIOCAD.

Спонсор конкурса — компания SkyGen: передовой дистрибьютор продукции для life science на российском рынке.
Спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
«Книжный» спонсор конкурса — «Альпина нон-фикшн»
Трехмерная структура хроматина одиночных клеток
О том, как геномная ДНК уложена в трехмерном пространстве ядра, известно уже немало. Во многом в этом помогла разобраться отлаженная техника фиксации конформации хромосом (chromosome conformation capture), наиболее производительный вариант которой называется Hi-C. Этот метод позволяет получить информацию о контактах разных участков ДНК друг с другом. Благодаря этому и целому ряду схожих методов, мы знаем о существовании топологически ассоциированных доменов (ТАДов), компартментов, петель , [1], [2]. Тем не менее на ряд вопросов о формировании и укладке структуры хроматина мы всё еще не можем ответить. Существенная проблема в том, что приходится только догадываться, как они устроены на физическом уровне.
Петли (loops) — неоднозначный термин в области биологии хроматина. В данном случае мы используем его как синоним пиков (peaks), которые видны на Hi-C-картах как яркие сигналы вне главной диагонали [3].
Отчасти причина в том, что Hi-C — это метод высокопроизводительного секвенирования, для которого требуется много ДНК: несколько десятков микрограммов. Чтобы получить такое количество ДНК, ученые проводят эксперимент на миллионах клеток. К сожалению, при этом не получается учесть, что каждая клетка и набор ее контактов ДНК — уникальны. Трехмерная организация хроматина одиночного ядра является следствием индивидуального процесса упаковки длинной молекулы ДНК под влиянием термодинамического движения молекул и никогда не повторяется. Глядя на миллионы прочтений после секвенирования, мы не можем назвать те контакты, которые встретились в одном ядре одновременно.
Прочтения, или риды (reads), — это последовательности нуклеотидов, которые можно получить из образца ДНК с помощью секвенирования [4]. Хотя существуют разнообразные методы секвенирования [5], в этой работе мы использовали один из самых эффективных и популярных: секвенирование нового поколения с помощью Illumina. Это высокопроизводительный метод, позволяющий прочитать сотни гигабайт последовательностей ДНК. Именно с этих данных начинается вычислительная работа: прочтения картируют и ищут в них контакты ДНК.
Более того, нам важно не только узнать набор контактов ДНК, но и реконструировать те физические процессы, которые, возможно, привели к тому, что сформировался именно такой набор контактов. К сожалению, такая реконструкция становится сложной и дает неоднозначные и трудно интерпретируемые результаты, если неизвестно, какие контакты ДНК имеются в одной клетке. Чтобы решить эту проблему, удалось интегрировать два экспериментальных протокола. Часть была взята от методов секвенирования геномов одиночных клеток [6], а часть — от Hi-C, и в итоге получилась техника single-cell Hi-C, или Hi-C одиночных клеток (рис. 1) [7].

Рисунок 1. Протокол Hi-C одиночных клеток. В Hi-C одиночных клеток первые этапы напоминают традиционное Hi-C на популяции: хроматин фиксируется, фрагментируется и сшивается с помощью лигазы. На следующем этапе, важном именно для данного протокола, каждая клеточка попадает в собственную лунку с помощью клеточного сортера (FACS). В каждой лунке оказывается порядка одного пикограмма ДНК. Этого недостаточно для того, чтобы прочитать ее последовательность с помощью методов высокопроизводительного секвенирования! Проблему решает амплификация с помощью специальной процессивной полимеразы фага phi29, которая значительно увеличивает количество копий ДНК. Такую амплифицированную ДНК уже можно отправлять на секвенирование и начинать обработку массивных данных.
Сшивки ДНК одиночных клеток
В казалось бы простом экспериментальном протоколе нашлось место и «подводным камням». Оказалось, что полимераза Phi29, с помощью которой мы амплифицировали геномную ДНК индивидуальных клеток, может совершать случайные «прыжки» между молекулами ДНК (рис. 2). Это приводит к возникновению искусственных сшивок, которые традиционные алгоритмы обработки данных Hi-С не отличают от настоящих контактов, образовавшихся в ядре клетки.

Рисунок 2. Последовательность манипуляций с ДНК в протоколе Hi-C одиночных клеток.
- Чтобы хроматин не развалился в процессе манипуляций в пробирке, контакты ДНК—ДНК и ДНК—белок «закрепляют» с помощью фиксации формальдегидом в самом начале эксперимента.
- Рестриктаза фрагментирует ДНК, но не в случайных местах. Каждая рестриктаза специфична к определенной последовательности нуклеотидов. Такая последовательность встречается в геноме тысячи раз, и по этим позициям и происходит разрезание. В данном случае использовался фермент DpnII, который делает надрез в ДНК перед последовательностью GATC.
- Лигаза образует сшивку между концами двух фрагментов. С большой вероятностью сшивка случится между разными участками генома, которые находились близко в пространстве, иначе говоря, контактировали. В результате образуется длинная непрерывная молекула ДНК, сформированная из пары или нескольких фрагментов, последовательно сшитых друг с другом.
- Полногеномная амплификация с помощью полимеразы Phi29.
- Прыжок полимеразы Phi29 между молекулами — типичная проблема Hi-C одиночных клеток с полимеразой фага Phi29.
иллюстрация Михаила Гурьева
К счастью, нашелся способ отличить настоящие и фальшивые контакты. Дело в том, что при лигировании соединение фрагментов происходит только по сайтам рестрикции, которые легко можно найти в последовательности ДНК. Прыжки полимеразы, напротив, происходят случайно и не имеют характерной подписи.
После картирования отдельно взятого прочтения Hi-C можно обнаружить стык лигирования и проверить, что он совпадает с сайтом рестрикции: значит, такому контакту можно доверять, с большой вероятностью он произошел в результате пространственного сближения и лигирования фрагментов хроматина внутри ядра. А по позициям картирования этих участков на геноме можно определить, какие участки ДНК последовательно соединялись в ходе эксперимента.
Чтобы вычислительно реализовать эту процедуру обработки данных, мы разработали алгоритм ORBITA (One-Read Based InTeractions Annotation), основанный на программе pairtools (рис. 3) [8]. Он позволяет извлекать и фильтровать настоящие сшивки ДНК, которые произошли в ходе пространственно-зависимого лигирования в ядре. Масштабное сравнение с подходом, который использовался ранее [9], [10], показало, что фильтрация по сайтам рестрикции помогает находить достоверные контакты Hi-C одиночных клеток и улучшает анализ.

Рисунок 3. ORBITA-подход для получения настоящих контактов Hi-C одиночных клеток
Совпадение или правило?
После того, как отлажена техника работы с Hi-C одиночных клеток, можно перейти к самому интересному — наблюдениям за новыми свойствами и механизмами формирования пространственной структуры хроматина. Пожалуй, самый интересный вопрос — являются ли механизмы и способы укладки хроматина разных организмов универсальными? Ранее Hi-C одиночных клеток проводили только для млекопитающих. Одно из важных наблюдений состояло в том, что хроматин одиночных клеток образует разбросанные по геному домены (участки с повышенной частотой контактов внутри).
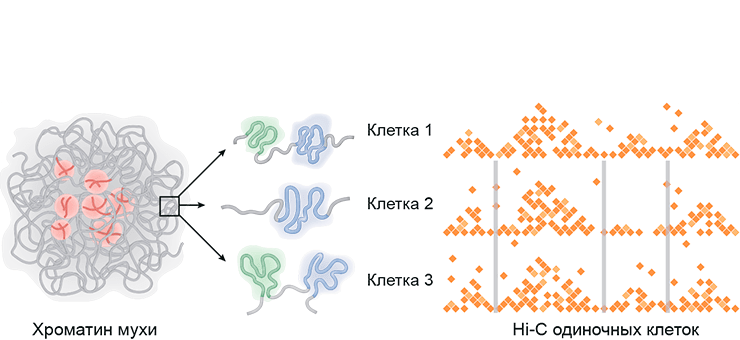
Теперь же мы применили Hi-C для одиночных клеток плодовой мушки дрозофилы [11]. Оказалось, что хроматин и этого организма в каждой отдельно взятой клетке сформирован из топологически ассоциированных доменов (ТАДов), однако структуры эти гораздо более упорядочены, чем у млекопитающих (рис. 4).

Рисунок 4. Сравнение результатов «популяционного» и «одноклеточного» Hi-C. Регион генома на традиционном популяционном Hi-C (верхняя панель), в объединении контактов 20 клеток (вторая панель) и в нескольких индивидуальных клетках дрозофилы. ТАДы видны как увеличенное количество контактов на участке («треугольники»). Положение ТАДов в индивидуальных клетках часто совпадает, а значит, что ТАДы — это довольно стабильные структурные единицы хроматина дрозофилы.
Конечно, совпадение позиций ТАДов может оказаться случайным, без какой-либо биологической причины. В данном случае с помощью моделирования и статистических тестов мы доказали, что относительно консервативные в разных клетках позиции ТАДов дрозофилы действительно являются следствием какого-то биологического механизма.
Каким может быть этот механизм? На наш взгляд, возможны как минимум два сценария (рис. 5). Хроматин дрозофилы может обладать особенными свойствами липкости, которые приводят к образованию стабильных конгломератов ДНК. Альтернативное объяснение — механизм выпетливания ДНК с помощью специальных факторов экструзии, которые останавливаются на участках ДНК, где располагаются активные гены. Обе эти гипотезы согласуются с тем, что известно о хроматине эукариот, однако для проверки этих гипотез требуются более детальные исследования (и, возможно, с помощью других методов).

Рисунок 5. Гипотезы формирования стабильных доменов в хроматине дрозофилы
Многолетняя коллаборация
Все эти исследования выполнялись в многолетней коллаборации, объединившей усилия ученых из Сколтеха, Института биологии гена РАН и МГУ им. М.В. Ломоносова, Национального центра научных исследований Франции, российско-французского Центра Понселе и нескольких других организаций.
Каждый из участников взял на себя отдельную важную роль, без которой проект не смог бы состояться. Так, Сергей Ульянов и Влада Захарова провели отработку экспериментальной техники. Моя роль заключалась в разработке вычислительных подходов и интерпретации данных. Павел Кос создал метод моделирования структуры хроматина единичных клеток дрозофилы. Кирилл Половников показал, что данные действительно отличаются от шума. Часть экспериментов провел наш давний коллаборатор Илья Флямер, который ранее применял подобную технику для исследования структуры хроматина ооцитов и зигот мыши [9], [10].
Похожий подход можно масштабировать и исследовать с его помощью разнообразие архитектур хроматина одиночных клеток, тканей и даже целых организмов. А пока что нам удалось зафиксировать неуловимую архитектуру хроматина клеток плодовой мушки и сформулировать гипотезы об упаковке ДНК этого организма. Подробности можно прочитать в журнале Nature Communications, где недавно вышла публикация о работе [12].
Работа поддержана грантами РФФИ №19-34-90136 и РНФ №19-14-00016.
Литература
- Организовать геном: запутанная история гипотез и экспериментов;
- Новый взгляд на геном: не просто цепочка генов, а трехмерная сеть, интегрирующая функциональные домены ядра;
- Suhas S.P. Rao, Miriam H. Huntley, Neva C. Durand, Elena K. Stamenova, Ivan D. Bochkov, et. al.. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 159, 1665-1680;
- Код жизни: прочесть не значит понять;
- 12 методов в картинках: секвенирование нуклеиновых кислот;
- Секвенирование единичных клеток (версия — Metazoa);
- Takashi Nagano, Yaniv Lubling, Tim J. Stevens, Stefan Schoenfelder, Eitan Yaffe, et. al.. (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502, 59-64;
- Parsing sequence alignments into Hi-C pairs. (2020). GitHub;
- Половая жизнь хроматина;
- Ilya M. Flyamer, Johanna Gassler, Maxim Imakaev, Hugo B. Brandão, Sergey V. Ulianov, et. al.. (2017). Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 544, 110-114;
- Модельные организмы: дрозофила;
- Sergey V. Ulianov, Vlada V. Zakharova, Aleksandra A. Galitsyna, Pavel I. Kos, Kirill E. Polovnikov, et. al.. (2021). Order and stochasticity in the folding of individual Drosophila genomes. Nat Commun. 12.



Комментарии
0Чтобы оставить комментарий, необходимо
войти