Рожденные стареть
14 января 2025
Рожденные стареть
- 1059
- 0
- 6
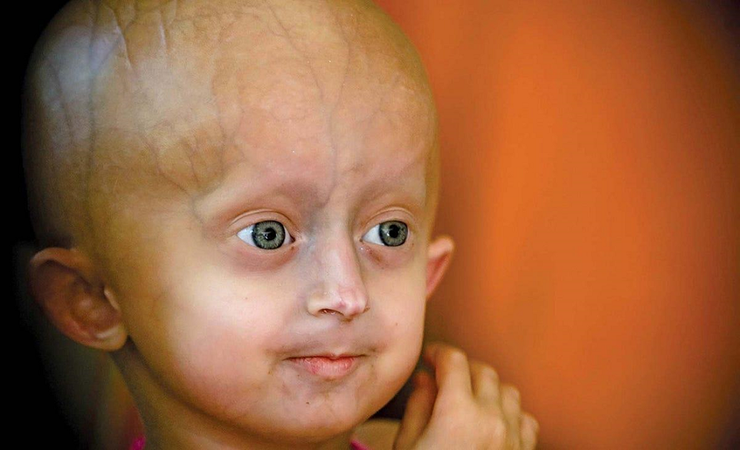
Ребенок с синдромом прогерии Хатчинсона-Гилфорда.
Рисунок в полном размере.
-
Автор
-
Редакторы
Статья на конкурс «Био/Мол/Текст»: Помните ли вы фантастическую историю Ф. Скотта Фицджеральда про необычного человека по имени Бенджамин Баттон, ставшую впоследствии основой фильма «Загадочная история Бенджамина Баттона»? Это история о человеке, который родился восьмидесятилетним мужчиной и в течение последующих восьмидесяти лет физиологически развивался в обратном порядке. Несмотря на фантастичность, в этой новелле есть момент реалистичности — главный герой рождается пожилым, что очень резонирует с редким генетическим заболеванием, при котором человек стареет преждевременно и молниеносно — детской прогерией (синдромом Хатчинсона-Гилфорда). По сравнению с другими прогероидными синдромами, которые различаются в зависимости от мутировавшего гена, при этом типе прогерии признаки преждевременного старения проявляются наиболее ярко. Для детей с этим синдромом жизнь настолько коротка, что они уже к трем годам приобретают вид 60-летнего человека и умирают, едва достигнув подросткового возраста. Прогерия изучается исследователями не только для того, чтобы найти заветное лекарство для помощи стареющим детям, но и для того, чтобы открыть путь к лечению миллионов взрослых с сердечно-сосудистыми заболеваниями и инсультом, связанными с естественным процессом старения. В этой статье речь пойдет о том, что такое детская прогерия и каков механизм ее развития, а также будут рассмотрены новейшие терапевтические достижения и будущие перспективы. Но обо всем по порядку.

Конкурс «Био/Мол/Текст»-2024/2025
 Эта статья опубликована в номинации «Старение и долголетие» конкурса «Био/Мол/Текст»-2024/2025 и заслужила приз Honoris causa.
Эта статья опубликована в номинации «Старение и долголетие» конкурса «Био/Мол/Текст»-2024/2025 и заслужила приз Honoris causa.
Генеральный партнер конкурса — международная инновационная биотехнологическая компания BIOCAD.
«Книжный» спонсор конкурса — «Альпина нон-фикшн»
Краткая история загадочной болезни
В далеком 1886 году британский врач, хирург, офтальмолог, дерматолог, венеролог и патолог Джонатан Хатчинсон впервые описал случай преждевременного старения у шестилетнего мальчика, которое проявлялось истончением кожных покровов и исчезновением подкожной клетчатки [1]. Чуть позже, в 1897 году похожий случай был описан хирургом Гастингсом Гилфордом (рис. 1). Из-за поразительного совпадения с нормальным старением он ввел термин «прогерия», который происходит от греческого и означает «преждевременно старый». Позже этот синдром стали называть синдромом прогерии Хатчинсона-Гилфорда (HGPS).

Рисунок 1А. Врач Джонатан Хатчинсон.

Рисунок 1Б. Врач Гастингс Гилфорд.
После того, как в конце XIX века болезнь была описана, никакого прогресса в изучении болезни не наблюдалось, за исключением все более подробного сбора симптомов, вплоть до конца XX века. Так, в 1972 году Ф.Л. ДеБаск сообщил о четырех новых случаях HGPS и составил первый подробный список случаев из литературы, похожих на прогерию. У всех своих пациентов с HGPS он обнаружил проблемы с сердечно-сосудистой системой [2].
В 1996 году в США в семье врачей родился ребенок с прогерией — Сэм Бернс (рис. 2). В 1999 году, когда Сэму было три года, его родители вместе с остальными членами семьи и друзьями основали Фонд исследований прогерии (PRF) с целью выявления причин прогерии и поиска способа вылечить своего сына и всех других детей с прогерией (на тот момент их было всего 45).

Рисунок 2. Сэм Бернс в возрасте 12 лет со своими родителями.
В 2000 году, благодаря картированию генома человека, исследование прогерии совершило решающий прорыв, который стал лишь первым шагом всех последующих исследований. Уже в 2003 году было обнаружено, что виновником прогерии является мутация в гене LMNA, кодирующем ламин А/С [3]. А в 2007 году, благодаря деятельности PRF, было организовано первое клиническое испытание, в ходе которого препараты (ингибиторы фарнезилтрансферазы, FTI) тестировались на детях с прогерией.
В 2013 году вышел документальный фильм «Жизнь с точки зрения Сэма», который повествует о запуске первого испытания препарата от прогерии с участием 26 детей из 16 стран (включая Сэма) и его последующих усилиях по публикации результатов в рецензируемом журнале. Сэм умер 10 января 2014 года в возрасте 17 лет, оставив после себя вдохновляющее наследие, которое теперь побуждает PRF и его сторонников продолжать поиски лекарства с огромной решимостью. За несколько месяцев до смерти Сэм Бернс прочел публичную лекцию о «философии своей счастливой жизни».
Уникальный почерк прогерии
По данным PRF, на 30 июня 2024 года во всем мире живет 154 ребенка с прогерией Хатчинсона—Гилфорда, независимо от пола и этнической принадлежности. Дети с HGPS выглядят обычными при рождении, однако уже в возрасте до одного года может наблюдаться задержка развития, потеря подкожного жира, выпадение волос, выступающие на голове вены и открытый родничок. Для детей с этим синдромом характерны отличительные внешние особенности — большая голова, узкая переносица, узкий кончик носа, тонкая красная каемка верхней и нижней губы, маленький рот и ретро- и микрогнатия (аномалия челюстно-лицевого скелета, которая характеризуется недоразвитыми челюстями) (рис. 3) [4].

Рисунок 3. Изменения лица, наблюдаемые у трех пациентов с прогерией с течением времени: (а), (б) — пациент 1 в возрасте 10 месяцев и (в) 3 года; (г) пациент 2 в возрасте 9 месяцев; (д) 18 месяцев и (е)5 лет; (ж) пациент 3 в возрасте 8 месяцев, (з) 2 года и (и) 6 лет.
Общими особенностями также являются короткие ключицы и общая тугоподвижность суставов. Более поздние признаки включают потерю слуха и скученность зубов. Все эти изменения приводят к характерному старческому виду. Но при всем этом чаще всего дети-старики имеют нормальный интеллект. Как правило, эти дети хорошо учатся, а также прекрасно осознают свое состояние. Как будто за их плечами — большой жизненный опыт.
Преждевременная смерть детей с прогерией обычно наступает во втором десятилетии жизни из-за сердечно-сосудистых осложнений [4]. Как и у любого человека, страдающего от болезни сердца, обычными событиями для детей с прогерией являются высокое давление, инсульты, стенокардия, увеличенное сердце и сердечная недостаточность — то есть все состояния, с которыми часто сталкиваются пожилые люди.
Так почему же эти дети стареют?
Ламин А
Прежде чем рассказать о причинах возникновения детской прогерии, стоит упомянуть о белках ламинах, из которых построены ядерные ламины [5]. Это промежуточные филаменты, состоящие из белков типа V — основные компоненты двухслойной ядерной оболочки, которая, в свою очередь, физически отделяет ядро от внешней среды и обеспечивает интерфейс для связи генома с этой средой [6]. Ламины участвуют в стабильности ядра и структуре хроматина [7], а также экспрессии многих генов [8]. У позвоночных животных и человека ламины бывают двух типов: A и B [9], [10]. Часто ламин типа А называют ламин А/С — из-за того, что мРНК ламина A и ламина C продуцируются одним и тем же геном LMNA посредством альтернативного сплайсинга, и, поскольку они довольно похожи (по первым 566 аминокислотам), их часто изучают вместе [8].
В то время как ламины типа B экспрессируются в целом во всех клетках, ламины типа A экспрессируются только в дифференцированных клетках, что, по-видимому, определяет специфические функции этого типа ламина в клетке. Более того, уровень экспрессии ламина A варьируется в разных тканях [11].
Ламин A содержит три структурных домена: центральный α-спиральный стержневой домен, короткий глобулярный аминоконцевой «головной» домен и длинный карбоксиконцевой «хвостовой» домен. В свою очередь, стержневой домен состоит из трех спиральных сегментов (1A, 1B и 2), соединенных короткими линкерами L1 и L12 (рис. 4) [12].

Рисунок 4. Структура ламина A. Центральный домен филамента разделен на субдомены 1A, 1B и 2, которые соединены линкерами L1 и L12. Хвостовой домен включает сигнал ядерной локализации (NLS), глобулярную иммуноглобулиновую складку (Ig-подобный домен) и мотив CaaX, где C — цистеин, a — алифатическая аминокислота, а X — любая другая аминокислота.
В нормальных условиях зрелый белок ламина А образуется из предшественника — преламина А, пройдя ряд этапов предварительной обработки. На первом этапе цистеин в C-концевом мотиве, обозначаемом как CaaX, модифицируется с помощью фермента фарнезилтрансферазы (FTase). На втором этапе последние три аминокислоты белка (т.е . —aaX ) «отрезаются» с помощью мембранных протеаз эндоплазматического ретикулума (ЭР) — ZMPSTE24 (или RCE1). Далее фарнезилцистеин метилируется мембранной метилтрансферазой ЭР (ICMT). Наконец, последние 15 аминокислот белка (включая С-концевой метиловый эфир фарнезилцистеина) отрезаются металлопротеазой ZMPSTE24, высвобождая зрелый ламин А [13].
Но бывает и так, что вырабатывается аномальная форма белка ламина А — о чем далее и пойдет речь.
Всего одна мутация
Ген LMNA является «горячей точкой» для мутаций, вызывающих заболевания, и привлекает к себе значительное внимание из-за своей связи с различными заболеваниями человека. Описано более 300 мутаций в гене LMNA, которые связаны с заболеваниями, известными под общим названием «ламинопатии».
Синдромы ламинопатий впервые были описаны еще в конце позапрошлого столетия, но только в 1999 году стало известно, что аутосомно-доминантная мышечная дистрофия Эмери-Дрейфуса (EDMD) связана с мутацией в гене ламина А/С [14]. Позже с ламиновыми мутациями были ассоциированы еще более десятка заболеваний, к числу которых относятся конечностно-поясная мышечная дистрофия 1В [15], мандибулоакральная дисплазия (MAD) [16], семейные липодистрофии [17], атипичный синдром Вернера [18] и другие.
Синдром прогерии Хатчинсона-Гилфорда также относится к ламинопатиям. В 90% случаев это заболевание вызывается точечной мутацией de novo в гене LMNA, которая обусловлена заменой цитозина на тимин в позиции 1824 нуклеотидной последовательности (c.1824C>T) (рис. 5) [19], [20].

Рисунок 5. Структура гена LMNA человека. Ламин C кодируется экзонами 1–10, а ламин A кодируется экзонами 1–12. Различные мутации связаны с патологиями, вызывающими прогероидные синдромы и/или заболевания, поражающие соединительные ткани.
EDMD — мышечная дистрофия Эмери—Дрейфуса. APS — атипичные прогероидные синдромы. HGPS — синдром прогерии Хатчинсона—Гилфорда. AWS — атипичный синдром Вернера. FPLD — семейная парциальная липодистрофия Даннигана. MAD — мандибулоакральная дисплазия.
Хотя эта мутация не должна нести за собой особых последствий, поскольку оба нуклеотидных триплета (гуанин-гуанин-цитозин и гуанин-гуанин-тимин) кодируют глицин (название мутации по полипептидной цепи — p.Gly608Gly), при этом активируется сайт сплайсинга в РНК LMNA, в результате чего из молекулы вырезается около 50 нуклеотидов и вырабатывается аномальная форма белка ламина А, называемая прогерином. В отличие от ламина А, прогерин делает ядро нестабильным, и эта клеточная нестабильность может приводить к целой совокупности патологических изменений, которые, в свою очередь, приводят к процессу преждевременного старения при прогерии.
При HGPS могут быть выполнены первые три этапа посттрансляционного созревания (то есть фарнезилирование, расщепление и метилирование), в то время как четвертый этап обработки не может быть завершен, поскольку мутация устраняет второй сайт расщепления, распознаваемый металлопротеазой ZMPSTE24 преламина А. Это приводит к постоянно фарнезилированной форме прогерина (рис. 6) [21]. Считается, что этот «неправильный» белок при HGPS лежит в основе прогрессирования фенотипа заболевания.
 и прогерина (справа)")
Рисунок 6. Биосинтез нормального зрелого ламина А (слева) и прогерина (справа). Посттранскрипционная обработка преламина А проходит четыре стадии реакции, прежде чем стать зрелым ламином А: фарнезилирование цистеина CaaX-бокса ферментом FTase, расщепление aaX металлопротеазой ZMPSTE24, карбоксилметилирование фарнезилированного цистеина с помощью изопренилцистеинкарбоксилметилтрансферазы (ICMT) и расщепление 15-ти концевых аминокислот с помощью ZMPSTE24.
Одним из отличительных признаков прогероидных клеток являются аберрантно сформированные ядра. Так, ядра клеток, экспрессирующих ламин А, имеют нормальную яйцевидную форму, тогда как ядра клеток, экспрессирующих необработанную форму преламина А, имеют аномальную форму (рис. 7).
 и здорового человека (слева)")
Рисунок 7. Клеточное ядро пациента с HGPS (справа) и здорового человека (слева).
Больше всего при HGPS страдают клетки, которые подвергаются высоким и постоянным механическим нагрузкам. К ним относятся костная ткань, кожа и сосудистые клетки, особенно гладкомышечные клетки в крупных артериях. Сосудистые клетки постоянно подвергаются воздействию различных механических напряжений от кровотока и внутрипросветного давления и, таким образом, наиболее чувствительны к возмущениям этих сил [23]. Поэтому основной причиной смерти при детской прогерии являются инфаркты и инсульты. Такая чувствительность к механическим нагрузкам связана, прежде всего, с тем, что дисфункциональный ламин А не способен поддерживать механические свойства клеточного ядра и делает его хрупким и восприимчивым к внешнему воздействию [23], [24].
Быстро стареющие мыши
Учитывая малое количество пациентов с HGPS во всем мире, существуют сложности в проведении продольных исследований и клинических испытаний. В связи с этим важным аспектом при изучении прогерии является использование животных моделей.
За последние десятилетия разработано несколько животных моделей HGPS для изучения самых разных аспектов заболевания. Первые из этих моделей были созданы путем изменения этапов посттрансляционной модификации ламина A. Так, в 2002 году две лаборатории разработали модель мыши Zmpste24 −/−, в которой отсутствует металлопротеаза ZMPSTE24, необходимая для расщепления карбоксильной группы преламина A на последнем этапе биосинтеза ламина A. Гомозиготные мыши Zmpste24 −/− накапливают необратимо модифицированный преламин А, лишены зрелого ламина А, в связи с чем у них развивается настоящий прогероидный фенотип, который включает дефекты мышц и костей, алопецию, липодистрофию и сокращение продолжительности жизни [25], [26]. Однако у этих мышей практически не наблюдалось серьезных сосудистых изменений, и потому научное сообщество нуждалось в других мышиных моделях, которые более точно напоминают патологию HGPS.
В 2006 году была создана мышиная модель, которая несет мутировавшую человеческую LMNA G608G на бактериальной искусственной хромосоме (BAC) — G608G BAC. Эта модель воспроизвела некоторые аспекты сосудистого фенотипа, связанного с HGPS [27], и потому представляет ценный инструмент для тестирования терапевтических подходов. Например, одно из исследований, проведенных на гомозиготных мышах G608G BAC показало, что лечение антисмысловыми олигонуклеотидами [28], нацеленными на LMNA, снижает уровень прогерина и увеличивает продолжительность жизни мышей (рис. 8) [29].
Пятью годами позже была создана мышиная модель Lmna G609G HGPS, в которой прогерин экспрессировался из-за альтернативного сплайсинга [30] гена LMNA. Эти мыши несут наиболее распространенное изменение у пациентов с HGPS — мутацию замены цитозина на тимин в позиции 1827 (p.G609G), которая эквивалентна мутации c.1824C>T (p.G608G) у людей [31].

Рисунок 8. Мыши с мутацией G608G BAC, которым вводили препарат внутривенно в хвостовую вену в дозе 60 мг/кг два раза в неделю, начиная с 2-недельного возраста. Слева: 7,5-месячная мышь, получавшая препарат. Справа: мышь такого же возраста, получавшая физраствор.
Хотя мышиные модели G608G BAC и Lmna G609G воспроизводят многие сосудистые изменения, обнаруженные у пациентов с HGPS, у них не развивается атеросклероз — одна из основных особенностей у пациентов с HGPS. Предполагается, что это связано с очень низким уровнем проатерогенных липопротеинов у мышей, что обеспечивает естественную устойчивость к развитию атеросклероза.
Чтобы преодолеть это ограничение, была создана мышиная модель Apoe −/− Lmna G609G/G609G [32]. Эти мыши несут мутацию LMNA, в результате которой образуется прогерин, и сконструированы таким образом, чтобы у них отсутствовал аполипопротеин E. Было продемонстрировано, что эта мутация вызывает атеросклероз у грызунов [33].
Исследования с использованием моделей мышей значительно продвинули знания о патологии, связанной с HGPS, включая сердечно-сосудистые заболевания, и проложили путь к разработке методов лечения, которые могут если не вылечить, то, по крайней мере, улучшить качество жизни детей с HGPS и увеличить ее продолжительность.
В поисках заветного лекарства
В настоящее время детская прогерия считается неизлечимым заболеванием. Однако уже сегодня достигнут определенный прогресс в разработке терапевтических средств, нацеленных на прекращение производства прогерина. Единственным препаратом, одобренным Управлением по контролю за продуктами и лекарствами (FDA) для лечения пациентов с HGPS, является лонафарниб, который является ингибитором фарнезилтрансферазы (FTI) [34].
Одним из авторов самой первой публикации, в которой сообщалось об эффективности лонафарниба, была мама Сэма Бернса — Лесли Б. Гордон, которая совместно с другими членами команды организовала это исследование и внесла непосильный вклад для того, чтобы оно было опубликовано [35]. Это исследование было проведено не совсем по правилам, так как обычно для проведения клинических исследований необходимо участие двух групп детей — экспериментальной, которая должна получать лекарство, и контрольной, которая получает плацебо. Но для небольшого числа детей с прогерией это было бы неправильно, если бы у одних детей был шанс продлить жизнь, а у других бы такого шанса не было. Поэтому ученые, понимая эту проблему, давали лонафарниб всем нуждающимся детям. Тем не менее, для доказательства увеличения продолжительности жизни у детей с прогерией ученые сопоставили детей в группе лечения с нелеченными детьми того же возраста и пола, родившимися во время лечения или после него.
В первом клиническом испытании препарата лонафарниба принимало участие двадцать шесть детей, средний возраст которых составлял 7 лет (рис. 9). Они получали лонафарниб в течение как минимум двух лет [35]. Один ребенок умер от инсульта через пять месяцев исследования (это не было связано с лечением), поэтому исход оценивался только для 25 участников. Результаты были обнадеживающими, так как около трети пациентов набрали вес после введения препарата. Кроме того, было отмечено улучшение здоровья сердечно-сосудистой системы и улучшение жесткости скелета и слуха. А дальнейшие исследования выявили увеличение средней продолжительности жизни на 1,6 года (рис. 9) [36].
")
Рисунок 9. Дети с прогерией, прошедшие лечение лонафарнибом (после двух лет клинических испытаний).
Недавние исследования показали, что более низкие уровни прогерина в крови отражают преимущество в выживании: чем дольше человек с прогерией остается на лонафарнибе, тем больше его польза. Уровни прогерина снижались примерно на 30–60% на протяжении всего времени приема препарата, а ожидаемая продолжительность жизни пациентов, находящихся на лечении более 10 лет, по оценкам, увеличилась почти на пять лет. Это увеличение средней продолжительности жизни более чем на 35%, с 14,5 лет до почти 20-летнего возраста! [37]
Основным механизмом действия лонафарниба является связывание фарнезилтрансферазы (FTase) с участком CaaX, что препятствуют способности FTase добавлять 15-углеродную группу к остатку цистеина CaaX-бокса и, соответственно, препятствует способности фермента взаимодействовать с прогерином [20]. Также было показано, что FTI улучшают подвижность теломер [38].
Затем была предложена тройная терапия — в дополнение к лонафарнибу в качестве лечения использовали также правастатин и золедронат. Правастатин используют, как правило, для профилактики заболеваний сердца, включая инфаркты и инсульты, а золедронат — для улучшения состояния костной ткани. Результаты клинического испытания такого тройного лечения показали улучшение плотности костной ткани по сравнению с монотерапией лонафарнибом [39].
В конце сентября 2024 года Фонд исследования прогерии заявил о начале клинического исследования препарата прогеринина — ингибитора связывания ламина А и прогерина. Ранее была показана эффективность данного препарата на мышах и отстутсвие токсичности для организма [40], [41]. Использование прогеринина позволило увеличить продолжительность жизни мышей на 14 недель, что в сравнении с лонафарнибом в семь раз выше.
Таким образом, было показано, что ингибиторы фарнезилтрансферазы эффективны в улучшении симптомов и снижении смертности у пациентов с HGPS, и сейчас основное внимание уделяется комбинированной терапии с FTI для выявления дальнейшего клинического улучшения.
Что касается доклинических исследований (в основном это исследования на мышах), то сейчас под прицелом у ученых имеется ряд препаратов с разным механизмом действия:
- ингибиторы изопренилцистеин-карбоксил-метилтрансферазы, которая во время посттрансляционных этапов обработки преламина А добавляет карбоксиметильную группу к терминальному цистеину. Ингибирование этого этапа может улучшить фенотипы заболевания, поскольку прогерин остается не только постоянно фарнезилированным, но и карбоксиметилированным. Было показано, что у мышей, которым вводили этот препарат, улучшились масса тела, сила хвата, диаметр поперечного сечения мышц и выживаемость [42];
- антисмысловые олигонуклеотиды (ASO), которые позволяют решить проблему альтернативного сплайсинга — основного механизма, который приводит к образованию прогерина. Недавно была создана библиотека потенциальных ASO для определения наиболее эффективных на основе изменений в уровнях мРНК ламина A, прогерина и ламина C [43];
- редакторы CRISPR-Cas9 [44] и адениновых оснований (ABE). Эти стратегии были разработаны для предотвращения экспрессии прогерина, воздействуя непосредственно на ген LMNA и его начальные продукты. CRISPR-Cas9 была разработана одновременно двумя независимыми группами, одна из которых использовала направляющую РНК (гРНК), нацеленную на экзон 11, в то время как другая использовала две гРНК, выбирая либо экзон 11, либо 12 [45], [46]. Доклинические исследования показали, что эта стратегия позволяет снизить уровни прогерина и улучшить ядерные дефекты в эмбриональных мышиных и человеческих фибробластах HGPS [47]. Мыши также продемонстрировали увеличение веса, улучшение выживаемости, фенотипического внешнего вида и мышечного захвата по сравнению с контрольными животными. Однако этот метод имеет и потенциальные ограничения, которые связаны, прежде всего, с высоким тропизмом вектора для печени, сердца и мышц, и низким тропизмом для аорты и легких.
Редакторы адениновых оснований преобразуют пары нуклеотидов A—T в C—G с использованием тРНК-зависимой аденозиндезаминазы [48], одиночной направляющей РНК (sgRNA) и измененной молекулы Cas9. Эффективность данной стратегии была показана как на фибробластах пациентов с HGPS, так и в мышиных моделях HGPS. Введение мышам лентивирусного вектора AAV9 [49] позволило увеличить их продолжительность жизни вдвое [50]. Таким образом, ABE представляют собой инновационную стратегию лечения детской прогерии с минимальным риском возникновения изменений в геноме. Дальнейшие исследования должны прояснить эффективность лечения, поскольку оно выглядит многообещающим, особенно с точки зрения продолжительности жизни.
Несмотря на то, что многие из этих методов лечения показали свою эффективность в улучшении фенотипов HGPS, они все же не являются лекарством. В ближайшем будущем решение для полного устранения клинических проявлений, вероятно, будет включать комбинации нескольких методов лечения.
Прогерин при физиологическом старении
На сегодняшний день нет однозначного ответа на вопрос о том, способствует ли выработка прогерина физиологическому старению. В 2006 году американские ученые Скаффиди и Мистели показали, что ядра в фибробластах кожи, полученных от пожилых людей, приобрели дефекты, похожие на дефекты в клетках детей с HGPS, включая изменения в модификациях гистонов и повышенное повреждение ДНК [46]. Вместе с тем, они обнаружили мРНК прогерина в фибробластах от здоровых пожилых людей, правда, на уровнях примерно в 50 раз более низких по сравнению с таковыми у пациентов с HGPS. Однако с возрастом уровни мРНК прогерина не увеличивались в нормальных фибробластах, оставляя открытым вопрос о том, способствует ли это старению. Позже было показано, что прогерин является биомаркером нормального клеточного старения, может быть потенциально связан со старением у пожилых людей [51] и играет роль в развитии сердечно-сосудистого старения в целом [52]. Таким образом, хотя и в гораздо меньших количествах, прогерин обнаруживается у людей, не страдающих прогерией, и поэтому может играть роль в естественном старении.
Заключение
Несмотря на то, что прогерия является редким заболеванием, она имеет важное сходство с физиологическим старением и, соответственно, может рассматриваться как ускоренный способ старения. Этот факт стимулирует исследовательские усилия, направленные на поиск лекарства от этой болезни, в надежде не только быть полезным для пациентов с прогерией, но и предоставить ценную информацию о возможности победы над самим процессом старения, что является одной из самых горячих тем современных исследований.
Нам, обычным условно здоровым людям, сложно представить, каково это — прожить такую короткую и малоприятную жизнь, как у детей с прогерией. Но, несмотря на многочисленные трудности, они способны вдохновлять не только ученых для поиска лекарства от старения, но и быть учителями для простых людей. Как бы то ни было, эти молниеносно стареющие дети невольно напоминают нам о неизбежности смерти и скоротечности жизни, в которой ценно каждое мгновение. 17-тилетний Сэм Бернс, выступая перед публикой незадолго до своей смерти, определил философию жизни основными правилами: смиритесь с тем, чего вы не можете сделать, а делайте то, что можете; окружите себя людьми, с которыми вам хочется быть; всегда продолжайте двигаться вперед. А последние слова, сказанные им тогда, подчеркивают его жажду к жизни: «Никогда не пропускайте вечеринку, если не можете удержаться. Завтра вечером в моей школе будет дискотека, и я там буду!».
Литература
- Jonathan Hutchinson. (1886). Congenital Absence of Hair and Mammary Glands with Atrophic Condition of the Skin and its Appendages in a Boy Whose Mother Had Been Almost Wholly Bald from Alopecia Areata from the Age of Six. Med Chir Trans.. MCT-69, 473-477;
- Franklin L. DeBusk. (1972). The Hutchinson-Gilford progeria syndrome. The Journal of Pediatrics. 80, 697-724;
- Maria Eriksson, W. Ted Brown, Leslie B. Gordon, Michael W. Glynn, Joel Singer, et. al.. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 423, 293-298;
- J. Mazereeuw-Hautier, L.C. Wilson, S. Mohammed, D. Smallwood, S. Shackleton, et. al.. (2007). Hutchinson–Gilford progeria syndrome: clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature. Br J Dermatol. 156, 1308-1314;
- Ядерная ламина и пространственная организация хроматина у дрозофилы;
- Stephen A. Adam. (2017). The Nucleoskeleton. Cold Spring Harb Perspect Biol. 9, a023556;
- Путешествие внутрь клеточного ядра, или Системная биология хроматина;
- Niina Dubik, Sabine Mai. (2020). Lamin A/C: Function in Normal and Tumor Cells. Cancers. 12, 3688;
- Travis A Dittmer, Tom Misteli. (2011). The lamin protein family. Genome Biol. 12, 222;
- Takeshi Shimi, Mark Kittisopikul, Joseph Tran, Anne E. Goldman, Stephen A. Adam, et. al.. (2015). Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. MBoC. 26, 4075-4086;
- Yosef Gruenbaum, Roland Foisner. (2015). Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu. Rev. Biochem.. 84, 131-164;
- Anna Malashicheva, Kseniya Perepelina. (2021). Diversity of Nuclear Lamin A/C Action as a Key to Tissue-Specific Regulation of Cellular Identity in Health and Disease. Front. Cell Dev. Biol.. 9;
- Catherine Coffinier, Hea-Jin Jung, Ziwei Li, Chika Nobumori, Ui Jeong Yun, et. al.. (2010). Direct Synthesis of Lamin A, Bypassing Prelamin A Processing, Causes Misshapen Nuclei in Fibroblasts but No Detectable Pathology in Mice. Journal of Biological Chemistry. 285, 20818-20826;
- Gisèle Bonne, Marina Raffaele Di Barletta, Shaida Varnous, Henri-Marc Bécane, El-Hadi Hammouda, et. al.. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 21, 285-288;
- A. Muchir. (2000). Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Human Molecular Genetics. 9, 1453-1459;
- Giuseppe Novelli, Antoine Muchir, Federica Sangiuolo, Anne Helbling-Leclerc, Maria Rosaria D’Apice, et. al.. (2002). Mandibuloacral Dysplasia Is Caused by a Mutation in LMNA-Encoding Lamin A/C. The American Journal of Human Genetics. 71, 426-431;
- H. Cao. (2000). Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Human Molecular Genetics. 9, 109-112;
- Lishan Chen, Lin Lee, Brian A Kudlow, Heloisa G Dos Santos, Olav Sletvold, et. al.. (2003). LMNA mutations in atypical Werner's syndrome. The Lancet. 362, 440-445;
- Aselah Lamis, Shiza W Siddiqui, Tejaswini Ashok, Nassar Patni, Mahejabeen Fatima, Asiff Nathi Aneef. (2022). Hutchinson-Gilford Progeria Syndrome: A Literature Review. Cureus;
- Noelle J. Batista, Sanket G. Desai, Alexis M. Perez, Alexa Finkelstein, Rachel Radigan, et. al.. (2023). The Molecular and Cellular Basis of Hutchinson–Gilford Progeria Syndrome and Potential Treatments. Genes. 14, 602;
- Janine Reunert, Rüdiger Wentzell, Michael Walter, Sibylle Jakubiczka, Martin Zenker, et. al.. (2012). Neonatal progeria: increased ratio of progerin to lamin A leads to progeria of the newborn. Eur J Hum Genet. 20, 933-937;
- Jon Macicior, Beatriz Marcos-Ramiro, Silvia Ortega-Gutiérrez. (2021). Small-Molecule Therapeutic Perspectives for the Treatment of Progeria. IJMS. 22, 7190;
- Kevin L. Shores, George A. Truskey. (2024). Mechanotransduction of the vasculature in Hutchinson-Gilford Progeria Syndrome. Front. Physiol.. 15;
- Melanie Maurer, Jan Lammerding. (2019). The Driving Force: Nuclear Mechanotransduction in Cellular Function, Fate, and Disease. Annu. Rev. Biomed. Eng.. 21, 443-468;
- Martin O. Bergo, Bryant Gavino, Jed Ross, Walter K. Schmidt, Christine Hong, et. al.. (2002). Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. U.S.A.. 99, 13049-13054;
- Alberto M. Pendás, Zhongjun Zhou, Juan Cadiñanos, José M.P. Freije, Jianming Wang, et. al.. (2002). Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase–deficient mice. Nat Genet. 31, 94-99;
- Renee Varga, Maria Eriksson, Michael R. Erdos, Michelle Olive, Ingrid Harten, et. al.. (2006). Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. U.S.A.. 103, 3250-3255;
- Есть ли смысл в антисенсах?;
- Michael R. Erdos, Wayne A. Cabral, Urraca L. Tavarez, Kan Cao, Jelena Gvozdenovic-Jeremic, et. al.. (2021). A targeted antisense therapeutic approach for Hutchinson–Gilford progeria syndrome. Nat Med. 27, 536-545;
- Белки против РНК — кто первым придумал сплайсинг?;
- Fernando G. Osorio, Claire L. Navarro, Juan Cadiñanos, Isabel C. López-Mejía, Pedro M. Quirós, et. al.. (2011). Splicing-Directed Therapy in a New Mouse Model of Human Accelerated Aging. Sci. Transl. Med.. 3;
- Magda R. Hamczyk, Ricardo Villa-Bellosta, Pilar Gonzalo, María J. Andrés-Manzano, Paula Nogales, et. al.. (2018). Vascular Smooth Muscle–Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation. 138, 266-282;
- Moritz von Scheidt, Yuqi Zhao, Zeyneb Kurt, Calvin Pan, Lingyao Zeng, et. al.. (2017). Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metabolism. 25, 248-261;
- Sohita Dhillon. (2021). Lonafarnib: First Approval. Drugs. 81, 283-289;
- Leslie B. Gordon, Monica E. Kleinman, David T. Miller, Donna S. Neuberg, Anita Giobbie-Hurder, et. al.. (2012). Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. U.S.A.. 109, 16666-16671;
- Leslie B. Gordon, Joe Massaro, Ralph B. D’Agostino, Susan E. Campbell, Joan Brazier, et. al.. (2014). Impact of Farnesylation Inhibitors on Survival in Hutchinson-Gilford Progeria Syndrome. Circulation. 130, 27-34;
- Leslie B. Gordon, Wendy Norris, Sarah Hamren, Robert Goodson, Jessica LeClair, et. al.. (2023). Plasma Progerin in Patients With Hutchinson-Gilford Progeria Syndrome: Immunoassay Development and Clinical Evaluation. Circulation. 147, 1734-1744;
- Winnok H. De Vos, Frederik Houben, Ron A. Hoebe, Raoul Hennekam, Baziel van Engelen, et. al.. (2010). Increased plasticity of the nuclear envelope and hypermobility of telomeres due to the loss of A–type lamins. Biochimica et Biophysica Acta (BBA) - General Subjects. 1800, 448-458;
- Leslie B. Gordon, Monica E. Kleinman, Joe Massaro, Ralph B. D’Agostino, Heather Shappell, et. al.. (2016). Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation. 134, 114-125;
- So-mi Kang, Min-Ho Yoon, Jinsook Ahn, Ji-Eun Kim, So Young Kim, et. al.. (2021). Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun Biol. 4;
- So-mi Kang, Seungwoon Seo, Eun Ju Song, Okhee Kweon, Ah-hyeon Jo, et. al.. (2023). Progerinin, an Inhibitor of Progerin, Alleviates Cardiac Abnormalities in a Model Mouse of Hutchinson–Gilford Progeria Syndrome. Cells. 12, 1232;
- Xue Chen, Haidong Yao, Muhammad Kashif, Gwladys Revêchon, Maria Eriksson, et. al.. (2021). A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife. 10;
- Madaiah Puttaraju, Michaela Jackson, Stephanie Klein, Asaf Shilo, C. Frank Bennett, et. al.. (2021). Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson–Gilford progeria syndrome. Nat Med. 27, 526-535;
- Просто о сложном: CRISPR/Cas;
- Ergin Beyret, Hsin-Kai Liao, Mako Yamamoto, Reyna Hernandez-Benitez, Yunpeng Fu, et. al.. (2019). Single-dose CRISPR–Cas9 therapy extends lifespan of mice with Hutchinson–Gilford progeria syndrome. Nat Med. 25, 419-422;
- Olaya Santiago-Fernández, Fernando G. Osorio, Víctor Quesada, Francisco Rodríguez, Sammy Basso, et. al.. (2019). Development of a CRISPR/Cas9-based therapy for Hutchinson–Gilford progeria syndrome. Nat Med. 25, 423-426;
- Daniel Whisenant, Kayeong Lim, Gwladys Revêchon, Haidong Yao, Martin O. Bergo, et. al.. (2022). Transient expression of an adenine base editor corrects the Hutchinson-Gilford progeria syndrome mutation and improves the skin phenotype in mice. Nat Commun. 13;
- Т9 для транскриптов: что такое редактирование РНК и зачем оно нужно?;
- Лентивирусные векторы: как они стали лучшими векторами для терапии ex vivo;
- Paola Scaffidi, Tom Misteli. (2006). Lamin A-Dependent Nuclear Defects in Human Aging. Science. 312, 1059-1063;
- Dayle McClintock, Desiree Ratner, Meepa Lokuge, David M. Owens, Leslie B. Gordon, et. al.. (2007). The Mutant Form of Lamin A that Causes Hutchinson-Gilford Progeria Is a Biomarker of Cellular Aging in Human Skin. PLoS ONE. 2, e1269;
- Michelle Olive, Ingrid Harten, Richard Mitchell, Jeanette K. Beers, Karima Djabali, et. al.. (2010). Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging. ATVB. 30, 2301-2309.



Комментарии
0Чтобы оставить комментарий, необходимо
войти