Генная терапия нейромоторных болезней
19 мая 2023
Генная терапия нейромоторных болезней
- 5220
- 0
- 10
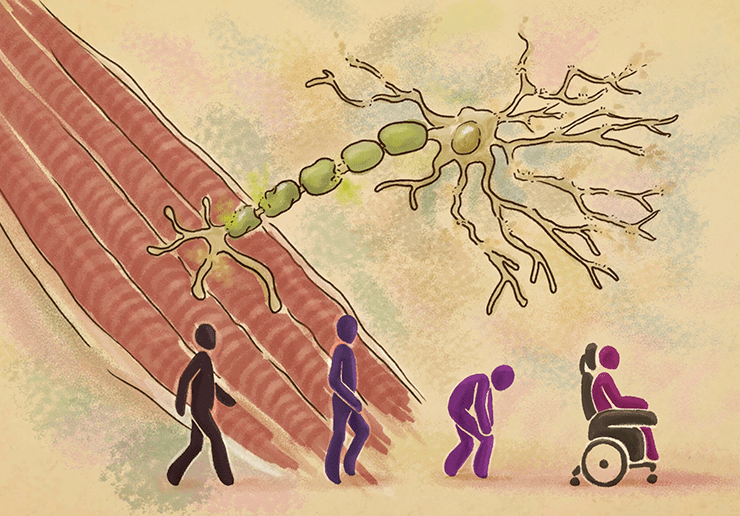
Мотонейроны иннервируют мышечные волокна, что позволяет осуществлять произвольные движения. При нейромоторных заболеваниях эта связь зачастую прервана, что приводит к широкому спектру патологических нарушений. Рисунок в полном размере.
иллюстрация Елены Беловой по 'Truly remarkable' drug helps motor neurone disease
-
Автор
-
Редакторы
-
Иллюстратор
Нервно-мышечные расстройства — «большая группа редких патологий, характеризующихся атрофией и слабостью скелетных мышц с частым поражением дыхательной и/или сердечной мускулатуры». Под это определение подпадают как болезни собственно мышц (дистрофии), так и заболевания двигательных (моторных) нейронов, в лечении которых на сегодня есть большая неудовлетворенная потребность. Надежду удовлетворить её связывают с генной терапией, ведь первопричины таких заболеваний зачастую кроются в наследственности (вызваны мутациями определенных генов). В этой статье спецпроекта о генной и клеточной терапиях обсудим перспективы их генного лечения.

Генная и клеточная терапии
Третий сезон спецпроекта о генной и клеточной терапиях рассказывает о конкретных применениях этих новейших методов для лечения нейромоторных, онкологических, офтальмологических и других заболеваний.
Партнер спецпроекта — Департамент разработки генотерапевтических препаратов одной из крупнейших российских биотехнологических компаний — BIOCAD. BIOCAD заслужил серьезные позиции на мировом фармацевтическом рынке благодаря выпуску лекарственных препаратов на основе антител.
Нейромоторные заболевания — группа нейродегенеративных состояний, отличающихся поражением двигательных (моторных) нейронов головного и/или спинного мозга. Зачастую эти нейроны просто дегенерируют, что приводит к мышечной слабости, потере способности передвигаться, хронической инвалидности и ранней смерти. Самые частые заболевания этой группы — боковой амиотрофический склероз (БАС), известный также как болезнь Лу Герига (по имени знаменитого бейсболиста, страдавшего от этой болезни, рис. 1); спинальная мышечная атрофия (СМА); и мышечная дистрофия Дюшенна (МДД) .
Основная причина дистрофии Дюшенна — именно мышечная патология — отличает её от других рассматриваемых в этой статье нейромоторных болезней, в патогенезе которых дегенерация мышц вторична по отношению к поражению двигательных нейронов и их корешков [1]. Строго говоря, МДД правильнее отнести к несколько более широкому классу нервно-мышечных расстройств (см. определение в аннотации выше).

В целом эти заболевания оказывают разрушительное воздействие на жизнь страдающих: они трудно поддаются лечению, поскольку вызывающие их причины пока невозможно полностью устранить — чаще всего речь идет о генетической патологии, которая влияет на организм с самого раннего возраста. К тому же патогенез зачастую варьирует от пациента к пациенту, что затрудняет выбор тактики лечения [2].
В некоторых случаях патология носит преимущественно врожденный характер (примерно в 1 случае из 10), но часто такие состояния возникают в результате сочетания (и взаимодействия) генетических факторов, негативных экологических воздействий (влияния окружающей среды) и образа жизни; при этом этиология бывает ясна не до конца [2].
Тем не менее сейчас уже изучен широкий спектр сугубо генетических дефектов, ассоциированных с патогенезом нейромоторных заболеваний, что привело к выработке различных способов компенсации их негативного действия. Генная терапия [3] представляет здесь особый интерес: ей подвластна регуляция работы генов, а значит — можно модулировать функцию затронутых болезнью белков с лечебными целями.
Для этого ныне разработаны различные стратегии: экспрессию генов меняют, чтобы прервать образование «патогенного белка», нивелировать (или свести к минимуму) мутационное повреждение (для «производства» функциональных белков), наконец (по классической схеме генной терапии) — доставить в клетку функциональный ген вместо поврежденного с использованием вектора-носителя. Последний подход, по-видимому, наиболее удачный — во всяком случае, работающая по такому принципу «Золгенсма» показывает ныне наиболее выдающиеся успехи (в лечении СМА, о чем поговорим ниже) [4].
Широкое применение для доставки лечебных генных конструкций имеют вирусные векторы (например, использованные в «Золгенсме» аденоассоциированные вирусы — AAV [5]), выбор которых, как мы увидим, также немаловажен.
Болезни и чем их лечить
Боковой амиотрофический склероз (БАС)
Основной особенностью заболевания считается избирательная гибель моторных нейронов в спинном мозге, стволе головного мозга и первичной моторной коре [6]. Сигнал до иннервируемых ими скелетных мышц перестает доходить должным образом, и они неминуемо ослабевают (происходит прогрессирующая атрофия). Поскольку эти клетки контролируют произвольные движения, это влечет за собой потерю подвижности, паралич, нарушения речи, и, в конечном итоге, смерть из-за дыхательной недостаточности (рис. 2).

Рисунок 2. Знаменитый астрофизик Стивен Хокинг страдал от БАС — эта болезнь парализовывала его долгие десятилетия, приведя в итоге к гибели от осложнений в возрасте 76 лет.
Эта болезнь впервые была описана еще в 1869 году, и сейчас уже известно, что она поражает примерно 5 человек на 100 000. Большинство случаев при этом не имеет четкой генетической причины (так называемые спорадические формы), хотя в 10% заболевание носит наследуемый характер [7].
На сегодняшний день идентифицировано более 140 различных мутаций, задействованных в наследственном патогенезе этой болезни, однако среди этого разнообразия есть и свои «лидеры» — дефекты генов, встречающиеся чаще других (они будут рассмотрены далее).
Мышечная дистрофия Дюшенна (МДД)
С распространенностью менее 10 случаев на 100 000 мужчин, МДД является тяжелым и изнурительным наследуемым заболеванием. Симптомы зачастую проявляются еще в детстве: в большинстве случаев страдает двигательное развитие — маленькие пациенты ходят медленно, не могут бегать; в дальнейшем функции моторики продолжают снижаться [6], [8], из-за необратимого разрушения мышечной ткани такие дети со временем утрачивают способность самостоятельно ходить. В конце концов совокупность дегенеративных изменений приводит к преждевременной смерти от респираторных или сердечных осложнений [8].
Этот страшный патогенез вызывается мутацией в гене, кодирующем дистрофин — белок цитоскелета скелетных и сердечных мышечных волокон. Этот белок очень важен для выживания мышечных клеток: его дисфункция из-за мутации собственно и приводит к некрозу мышечных клеток и прогрессирующей мышечной дегенерации со всеми вытекающими (рис. 3) [8].

Рисунок 3. Роль дистрофина в патогенезе МДД. Дистрофин играет роль своеобразного «якоря», соединяя актиновый цитоскелет с ассоциированным гликопротеиновым комплексом и стабилизируя тем самым клеточную мембрану мышечной клетки (сарколемму), что обеспечивает устойчивость и стабилизацию мышечного волокна при сокращении. Дисфункциональный дистрофин при МДД приводит к нестабильности сарколеммы — она разрушается, в результате содержимое мышечных клеток выходит в кровяное русло, что приводит к их гибели и, как следствие — к прогрессирующей мышечной дегенерации.
иллюстрация Елены Беловой по [2]
Спинальная мышечная атрофия (СМА)
Это основная наследственная причина младенческой смертности — распространенность этой болезни составляет около 1 случая на 6000 – 11 000 новорожденных [9].
Заболевание вызывается главным образом мутациями в гене SMN1 — одном из двух, кодирующих белок выживаемости двигательных нейронов SMN — Survival of motor neuron (название говорит само за себя — без него нейроны действительно погибают).
Другой ген, кодирующий белок SMN, — SMN2 — почти идентичная копия SMN1, но он не вовлечен в патогенез болезни большинства пациентов со СМА [8]. Правда и компенсировать «недостачу» функционального белка выживаемости мотонейронов ген второго типа, к сожалению, не способен — разница всего в один нуклеотид (по сравнению с SMN1) при экспрессии приводит к образованию нестабильного, плохо работающего белка SMN [8], [10].
С клинической точки зрения СМА характеризуется гибелью спинальных (нижних) двигательных нейронов, что постепенно приводит к мышечной слабости, прогрессирующему снижению двигательной функции, параличу и смерти от дыхательной недостаточности.
Эту болезнь в зависимости от тяжести подразделяют на четыре типа. К самой тяжелой спинальной мышечной атрофии 1 типа относят 60% больных: когда нет лечения, дети с этой формой не могут сидеть без посторонней помощи, и 90% из них умирают в возрасте до 20 месяцев [8].
О других типах СМА, а также более подробно об их патогенезе читайте в материале «Биомолекулы»: «Надежда для СМАйликов» [10].
Лечение
На сегодняшний день не существует средств, способных радикально и полностью вылечить нейромоторные заболевания. Применяемое лечение в основном облегчает симптомы (снятие спазмов миорелаксантами, облегчение боли противовоспалительными и др.), и лишь несколько препаратов эффективно замедляют развитие заболеваний [6]. Ниже обсуждаем некоторые из них.
Как сегодня лечат СМА?
«Золгенсма» — истинный прорыв
«Золгенсма» — препарат генной терапии, одобренный в США для лечения детей со СМА младше двух лет [4], а также относительно недавно и в России [11]. Это классическая геннозаместительная терапия: векторы AAV9 (о векторах подробнее рассказано во врезке ниже), несущие ген, кодирующий белок SMN1, вводят однократно и внутривенно в системный кровоток. Там они переносятся с кровью в ткани периферической и центральной нервной систем, проникая через гематоэнцефалический барьер и достигая ядер двигательных нейронов-мишеней в передних рогах спинного мозга (рис. 4).

Рисунок 4. Внутреннее строение спинного мозга. 1 — задние рога спинного мозга; 2 — боковые рога; 3 — передние рога; 4 — центральный канал; 5 — центральное промежуточное вещество; 6 — задняя центральная борозда; 7 — задняя боковая борозда; 8 — передняя центральная щель; 9 — задние канатики; 10 — боковой канатик; 11 — передний канатик.
Трансдуцируя моторные нейроны, AAV9 вносит в их ядра полностью функциональную копию гена SMN1, экспрессия которого и дает этим клеткам альтернативный источник важного для выживания белка SMN. За счет этого компенсируется его наследственное отсутствие — первопричина спинальной мышечной атрофии корректируется, что и приносит лечебный эффект [4].
Эффективность «Золгенсмы» подтверждена рядом клинических испытаний, старт которым был дан еще в 2014 году, и потому сейчас уже есть данные о действенности препарата спустя продолжительное время после его однократного введения.
Например, согласно исследованию NCT04042025, 10 пациентов, получивших высокие дозы AAV9 в самом первом испытании START, живы до сих пор и не нуждаются в искусственной вентиляции легких более чем через шесть лет после начала лечения [12]. Все «двигательные показатели», достигнутые в этом «дебютном» исследовании, также сохранены, что говорит о долгоиграющей эффективности лекарства.
Абстрагируясь от сухих данных клинических исследований, отметим, что «Золгенсма» в каком-то смысле способна на чудо: зафиксированы даже единичные случаи практически полного восстановления двигательных навыков (впрочем, чаще результат намного более скромный) [11], [12], и это при том, что в отсутствии лечения утрата способности ходить почти неминуема, да и смертность крайне высока [10].
Ложка дегтя
Несмотря на одобрение «Золгенсмы» и ее высокую эффективность в лечении СМА, существуют и связанные с ней риски. Компания-производитель сообщала о смерти двух детей, пролеченных «Золгенсмой», от острой печеночной недостаточности, что вновь привлекло внимание к безопасности этого препарата [8].
Токсичность для печени является одним из наиболее распространенных побочных эффектов AAV-терапии (при введении в системный кровоток эти векторы попадают в основном как раз в печень), а потому лекарства этого типа, к сожалению, безвредными не назовешь. Однако поскольку зачастую альтернативой такому лечению служит почти неминуемая гибель, риски эти, как ни странно, бывают вполне оправданы.
Некоторую надежду на лечение нейромоторных и других редких заболеваний дают также антисенсы — этот класс препаратов формально не относится к генной терапии (потому что это относительно небольшие молекулы), но представляет собой относительно новый способ воздействия на генетические механизмы клетки (подробнее во врезке ниже).
Генная терапия против БАС
Нацеливание на SOD1 и C9orf72
SOD1 — ген, кодирующий антиоксидантный фермент супероксиддисмутазу-1, и одновременно — популярная мишень в лечении БАС из-за частого возникновения в нем повреждающих мутаций, а также хорошо изученной их роли в инициации патогенеза этого заболевания [22].
Мутации этого гена обнаружили впервые еще в 1993 году, и сейчас уже известно, что они отвечают за 12–20% наследственных случаев БАС во всем мире; а в Азии мутация SOD1 и вовсе наиболее частая генетическая причина этого заболевания.
В норме супероксиддисмутаза-1 нейтрализует активные формы кислорода в цитоплазме и митохондриях, оказывая за счет этого свое нейропротективное действие. Однако при мутации белок начинает агрегировать, причем агрегаты его накапливаются в двигательных нейронах спинного мозга, что связывают с токсичностью, хотя точный механизм такого патогенеза ясен не полностью. По всей видимости, мутации в SOD1 приводят к нейродегенерации через совокупность деструктивных изменений: повышения окислительного стресса, деградации клеточных белков, воспаления с участием микроглии, токсической агрегации мутантных супероксиддисмутаз [23], дисфункции митохондрий и олигодендроцитов и др. [22].
Другой значимой причиной БАС, обусловливающей до 35–45% наследственных случаев этого заболевания, является экспансия — увеличение количества так называемых гексануклеотидных повторов (шестибуквенные последовательности типа GGGGCC) в гене C9orf72 ввиду затрагивающей его мутации [22]. Точного понимания особенностей молекулярного патогенеза при этом также пока нет — вопрос этот остается во многом открытым. Предполагается, что мутация C9orf72 как-то связана с нарушением ядерно-цитоплазматического транспорта в нейронах, что приводит к нарушению их функциональности и повреждению (со всеми вытекающими из этого последствиями) [22], [24], [25].
Так или иначе, сегодня мутации SOD1 и C9orf72 — основные мишени для генного лечения БАС, и для терапевтического воздействия на них предложены и апробированы несколько подходов.
Векторы и CRISPR
Аденоассоциированные векторы уже неоднократно использовали в попытках создать эффективную генную терапию против БАС, при этом самые разные стратегии в этой области разрабатываются и сегодня.
Например, такими носителями направляли короткие интерферирующие РНК для ингибирования образования мутантного белка SOD1 за счет нарушения этими молекулами его биосинтеза; доставляли и антисенсы (см. вставку выше) для такого модулирования сплайсинга, за счет которого при считывании информации с пре-мРНК пропускался бы экзон-2 (подход exon skipping), что приводило к укороченному транскрипту, разрушаемому в итоге естественными механизмами внутриклеточной деградации (это также прерывало биосинтез SOD1 [26]) и т.д.
На изолированных клетках, мышах и дрозофилах пробовали даже доставку широко известной системы CRISPR, что позволило отредактировать целевой ген и дало свой положительный эффект (табл. 1), продемонстрировав принципиальную возможность использования данного метода у человека. Впрочем, нужно оговориться, что в данных исследованиях имели место и «нецелевые события» (такие как многократное увеличение индел-мутаций в редактируемом гене [27]), что указывает на еще недостаточную проработанность применяемой методики, а значит, и потенциальные проблемы с безопасностью.
Что-то подобное можно сказать и о других методах: в доклинических испытаниях получаются многообещающие результаты, но поскольку клинических исследований пока немного (да и результаты их довольно-таки скромные [28]), говорить о серьезных прорывах немного преждевременно [26]. Имеющиеся на сегодняшний день достижения обобщены в таблице 1.
| Тип генной терапии | Модель | Основные результаты |
|---|---|---|
| AAV, доставляющие интерферирующую РНК | ||
| Ингибирование мутантного SOD1 | Линия мышей, экспрессирующих мутантную форму гена SOD1 (SOD1G93A мыши) | Отсроченный дебют БАС позволил временно сберечь двигательные нейроны и увеличить за счет этого продолжительность жизни мышей [30] |
| Улучшение выживаемости и функциональности моторных нейронов [31], [32] | ||
| Улучшение двигательной функции, увеличение мышечной массы и увеличение продолжительности жизни у мышей [33] | ||
| Увеличение средней продолжительности жизни мышей на 21% и сохранение у них моторной и дыхательной функций [34] | ||
| Повышение выживаемости тестируемых мышей и предотвращение у них ухудшения нервно-мышечной функции [26] | ||
| Человек; клинические исследования фазы I пациентов с БАС, вызванным мутацией гена SOD1 | У одного пациента на фоне улучшения состояния до стабильного уровня также снизился уровень SOD1 в спинном мозге. Второй пациент без клинических улучшений [35] | |
| Улучшение нервно-мышечной иннервации | ||
| Доставка AAV-векторами гена DOK7 (его экспрессия активирует киназу MuSK, ингибирующую деградацию окончаний двигательных нервов в нервно-мышечных соединениях) | SOD1G93A мыши | Улучшение двигательной активности и увеличение продолжительности жизни испытуемых мышей, но отсутствует влияние на количество двигательных нейронов [36] |
| Улучшение нейротрофики | ||
| Доставка AAV-векторами гена, кодирующего человеческий инсулиноподобный фактор роста 1 (hIGF1) (используется в качестве нейротрофического фактора) | SOD1G93A мыши | Значительно уменьшилась потеря двигательных нейронов переднего поясничного отдела спинного мозга, что приостановило атрофию мышц у тестируемых мышей. Это отсрочило начало болезни БАС и увеличило продолжительность их жизни [37] |
| AAV-CRISPR | ||
| Ингибирование мутантного SOD1 | SOD1G93A мыши | Снижение количества мутантного белка SOD1 больше чем в 2,5 раза в грудной и поясничной областях спинного мозга. На заключительном этапе у протестированных мышей было обнаружено примерно на 50% больше двигательных нейронов, и примерно на 37% было замедлено прогрессирование заболевания [38] |
| Делеция гена SOD1 и увеличение продолжительности жизни тестируемых мышей на 54,6% [39]. | ||
| Удаление мутаций из гена C9orf72 за счет его редактирования | Человеческие iPSCs (индуцированные плюрипотентные стволовые клетки) | Предотвращало генетические аномалии, связанные с мутацией, но не влияло на производство C9orf72 [40] |
| Дрозофила; человеческие iPSCs | Снижение активации апоптических путей и уменьшение очагов повреждения ДНК [41], [42] | |
Генная терапия мышечной дистрофии Дюшенна
Для лечения МДД логично постараться доставить ген дистрофина при помощи AAV, однако здесь ученые столкнулись с определенными трудностями. Дело в том, что дистрофин — один из самых больших генов человека: он включает 79 экзонов и 2,6 миллиона пар нуклеотидов, в одиночку (!) занимая около 0,1% всего генома. Неудивительно, что и приходящихся на такой крупный ген мутаций также немало — более 7000, включая делеции, охватывающие один или несколько экзонов, на которые приходится от 60 до 70% случаев заболеваний МДД [43].
Очевидно, подобрать годную генную терапию при таком патогенетическом разнообразии не так-то просто [44], однако сложность здесь не только (да и не столько) в этом. Эффективный перенос большого полноценного гена вместо поврежденного теоретически разрешил бы все проблемы, — но как его «впихнуть» в крошечный вектор (напомним: AAV имеют крайне ограниченную емкость) [5]?
Помимо этого дополнительную трудность составляет поражение при болезни почти всех мышц (скелетной, гладкой и сердечной мускулатуры, вместе составляющих ~40% общей массы тела у мужчин), что означает необходимость системной доставки большими дозами и влечет за собой издержки при дозировании [43]. А ведь векторы AAV вообще-то имеют узкое окно дозирования: их не должно быть слишком много — тогда может подключаться приобретенный иммунитет, и их действие нейтрализуется антителами (кроме того, совсем не стоит забывать о токсичности высоких доз векторов [34]); ну а маленькие дозы попросту не смогут обеспечить эффективность .
Подробнее о нюансах дозирования AAV, как и в целом про эти векторы можно узнать из статьи: «Время первых: как аденоассоциированные вирусы стали лучшими в доставке генов in vivo» [5].
Как же доставить дистрофин в клетку?
Решение проблемы чересчур крупного для доставки гена подсказала сама жизнь. Существует «разновидность» МДД — мышечная дистрофия Беккера со сходными симптомами и генным патогенезом, всё так же затрагивающим ген дистрофина. Только вот степень повреждения гена при этом заболевании менее серьезна, из-за чего и симптоматические проявления «мягче». Удивительно, но один из пациентов с этой болезнью даже прожил более 70 лет, хотя его ген дистрофина был «обрезан» почти наполовину. Это-то и натолкнуло исследователей на создание для лечения МДД миниатюризированной версии такого терапевтического гена — микро-/минидистрофина [43].
Доставку этого гена с использованием AAV в клинических исследованиях на данный момент тестируют три компании: Solid Biosciences, Pfizer и Sarepta Therapeutics (рис. 6).

Рисунок 6. Генетические конструкции для доставки уменьшенного дистрофина. Сверху полноразмерный дистрофин, ниже микродистрофины, разработанные компаниями Sarepta, Solid и Pfizer (слева также векторы, доставляющие данные гены) исследуемые в трех клинических испытаниях.
Sarepta Therapeutics
Sarepta уже успели пролечить четырех маленьких пациентов (в возрасте 4–6 лет) с использованием достаточно больших дозировок (2,0 × 1014 вг/кг — векторных геномов на килограмм). По результату: в тканях, отобранных с помощью биопсии, выявили, что примерно 81,2% мышечных волокон содержат микродистрофин, — ну а уровень его синтеза при этом 74,3%. Функциональные двигательные способности также улучшались, и побочные явления были минимальны.
Средний возраст этих пациентов сейчас уже старше девяти лет, а их двигательные показатели продолжили расти [43]. Успех этого исследования подтолкнул к продолжению испытаний, — на этот раз уже на 41 пациенте. В январе 2021 года были опубликованы первые его результаты за 48-недельный период: они продемонстрировали повышенное производство микродистрофина через 12 недель по сравнению с исходным уровнем (средний уровень биосинтеза увеличилась на 28,1%), и никаких новых опасений по поводу безопасности не возникло [43]. Позднее было запущено исследование NCT04626674 на 40 пациентах (в возрасте от трёх лет и старше), и если проанализировать все результаты, опубликованные Sarepta, окажется, что компания смогла закрепить свой успех.
Согласно комплексному анализу эффективности все три испытания (суммарно более 80 пролеченных пациентов) показали статистически значимое улучшение экспрессии гена функционального дистрофина и двигательных навыков в разные моменты времени: через один, два и более (до четырех) лет после начала лечения.
В частности, у всех получавших препарат спустя год отмечено улучшение результатов функциональных тестов по отношению к контролю: время подъема (Time-to-Rise) сократилось на 1,6 с, а время прохождения дистанции в 10 метров (10-meter walk/run) — на 0,7 с . Среднее же увеличение биосинтеза микродистрофина относительно исходного уровня по трем исследованиям за 12 недель наблюдения составило 55,7%.
Временные функциональные тесты измеряются в секундах. Уменьшение количества секунд для выполнения теста свидетельствует об улучшении двигательной функции (числа приведены по интегрированным данным трёх исследований).
В июле 2020 года FDA предоставило Sarepta статус Fast Track (для ускоренного рассмотрения и одобрения), а уже в ноябре 2022 года компания сообщила, что FDA приняло заявку на приоритетное рассмотрение (шесть месяцев вместо десяти) — это означает, что препарат становится всё ближе к выводу на рынок.
Solid Biosciences
Продукт Solid Biosciences вводился шести пациентам в возрасте от 4 до 17 лет в низких (5,0 × 1013 вг/кг) и более высоких (2,0 × 1014 вг/кг) дозировках. Было показано, что низкие дозировки малоэффективны: лишь около 10% мышечных волокон производили микродистрофин на уровне ниже 5%. В тоже время при высокой дозе от 10 до 70% мышечных волокон синтезировали этот белок на уровне 5–17,5% [43] (что в сравнении с данными Sarepta в общем-то тоже не особо впечатляет)
При этом наблюдались и серьезные нежелательные явления, приводившие даже к госпитализации (у одного пациента появилась почечная недостаточность, и ему потребовался кратковременный диализ). Сейчас все больные чувствуют себя хорошо. Из-за возникавших проблем компания дважды приостанавливала исследования, но сейчас снова их продолжает.
Pfizer
В исследовании продукта компании Pfizer изначально также испытывали как низкие (1,0 × 1014 вг/кг; для трех пациентов), так и более высокие (3,0 × 1014 вг/кг; для шести пациентов) дозы. Биосинтез микродистрофина предварительно оценивали через 2 месяца после начала лечения (первая контрольная точка), а затем — через 12 месяцев (вторая контрольная точка).
Согласно опубликованным данным, через 2 месяца после начала лечения у первой группы пациентов (низкое дозирование) производство микродистрофина повысилось на 20%, через 12 месяцев ее увеличение составило уже 24%. При более высоком дозировании в аналогичные временные промежутки уровень синтез повысился в первой контрольной точке на 35%, во второй — на 52%. У трех из пролеченных улучшились и двигательные функции [43].
К сожалению, более 40% больных страдали рвотой, тошнотой, снижением аппетита, повышением температуры. Были и более серьезные проблемы, даже потребовавшие неотложного вмешательства: постоянная рвота и обезвоживание организма с отказом почек [43], [45]. Эти случаи были эффективно пролечены и купированы в течение двух недель [43].
Pfizer сделали свою «работу над ошибками»: испытания приостановили, в протокол лечения внесли изменения для смягчения побочных эффектов; и в расширенной в дальнейшем до 19 человек когорте серьезных проблем больше почти не было (на 2020 год). Двигательные показатели пациентов улучшились, при этом лишь 30% из них испытали незначительные нежелательные явления [43].
Этот промежуточный успех позволил претендовать на большее: в октябре 2020 года продукт компании получил от FDA статус Fast Track, и затем ускоренное разрешение на начало испытаний III фазы клинических исследований. И всё бы ничего, но в дальнейшем серьезные нежелательные явления продолжились, апофеозом чего стала смерть одного из пациентов [43].
После этого FDA постановила прекратить клинические испытания; сейчас Pfizer расследует обстоятельства и причины смерти пациента, — ну а безопасность данного препарата (как и его возможное одобрение) теперь под большим вопросом.
Заглянем в будущее
Долгие годы обсуждаемые здесь болезни считались неизлечимыми, но, как показал случай с «Золгенсмой», успешно лечить их вполне можно — с помощью генной терапии. По крайней мере, когда генетический компонент является основным пусковым механизмом патогенеза.
Промежуточные результаты текущих доклинических и клинических испытаний указывают на потенциальную полезность самых разных стратегий лечения, что вселяет оптимизм в отношении будущих достижений и помощи страдающим такими заболеваниями людям.
В частности, многие исследовательские усилия сейчас сосредоточились на поиске новых терапевтических технологий, таких как редактирование генов системами типа CRISPR и нацеливание на различные РНК-мишени [29] (как для деградации транскриптов и прерывания биосинтеза, так и для модуляции сплайсинга с целью «исправляющей» генные дефекты модификации конечных белковых продуктов).
И хотя пока еще многие типы лечения остаются «сырыми» и, безусловно, нуждаются в дальнейшей доработке, совокупность различных исследовательских стратегий в будущем наверняка подарит нам еще не одну эффективную терапию.
Литература
- Nertiyan Elangkovan, George Dickson. (2021). Gene Therapy for Duchenne Muscular Dystrophy. JND. 8, S303-S316;
- Wei Chiu, Ya-Hsin Hsun, Kao-Jung Chang, Aliaksandr A. Yarmishyn, Yu-Jer Hsiao, et. al.. (2020). Current Genetic Survey and Potential Gene-Targeting Therapeutics for Neuromuscular Diseases. IJMS. 21, 9589;
- Генная терапия: познакомьтесь с лекарствами будущего;
- Самое дорогостоящее лекарство в мире;
- Время первых: как аденоассоциированные вирусы стали лучшими в доставке генов in vivo;
- Myrsini Chamakioti, Nikolaos Karantzelis, Stavros Taraviras. (2022). Advanced Gene-Targeting Therapies for Motor Neuron Diseases and Muscular Dystrophies. IJMS. 23, 4824;
- Kaspar B.K., Foust K., Cleveland D.W. (2020). Products and methods for treatment of amyotrophic lateral sclerosis. Patent EP3039146B1;
- Jean-Baptiste Brunet de Courssou, Alexandra Durr, David Adams, Jean-Christophe Corvol, Louise-Laure Mariani. (2022). Antisense therapies in neurological diseases. Brain. 145, 816-831;
- Denise M. Kay, Colleen F. Stevens, April Parker, Carlos A. Saavedra-Matiz, Virginia Sack, et. al.. (2020). Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genetics in Medicine. 22, 1296-1302;
- Надежда для СМАйликов;
- Магия для маглов: волшебный мир Гарри Поттера ближе, чем мы думаем;
- Proud C. (2022). Gene therapy updates in neuromuscular diseases. CGTlive;
- Blood-brain barrier. Связующее между двумя мирами — кровеносной и центральной нервной системами;
- Крохотные курьеры: как аденоассоциированные вирусы спасают жизни;
- Лентивирусные векторы: как они стали лучшими векторами для терапии ex vivo;
- Есть ли смысл в антисенсах?;
- Shikha Thakur, Apurba Sinhari, Priti Jain, Hemant R. Jadhav. (2022). A perspective on oligonucleotide therapy: Approaches to patient customization. Front. Pharmacol.. 13;
- Ashok Verma. (2018). Neuromuscular diseases: Recent advances in antisense oligonucleotide therapy. Astrocyte. 5, 81;
- Eric W. Ottesen. (2017). ISS-N1 makes the first FDA-approved drug for spinal muscular atrophy. Translational Neuroscience. 8, 1-6;
- Timothy M. Miller, Merit E. Cudkowicz, Angela Genge, Pamela J. Shaw, Gen Sobue, et. al.. (2022). Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 387, 1099-1110;
- Shilpa Narayanan, Akshay Shanker, Tanvi Khera, Balachundhar Subramaniam. (2021). Neurofilament light: a narrative review on biomarker utility. Fac Rev. 10;
- Ton Fang, Goun Je, Peter Pacut, Kiandokht Keyhanian, Jeff Gao, Mehdi Ghasemi. (2022). Gene Therapy in Amyotrophic Lateral Sclerosis. Cells. 11, 2066;
- Микроглия: роль «иммунных» клеток центральной нервной системы в здоровом мозге и при нейродегенеративных заболеваниях;
- Mehdi Ghasemi, Kiandokht Keyhanian, Catherine Douthwright. (2021). Glial Cell Dysfunction in C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Cells. 10, 249;
- Brian D. Freibaum, Yubing Lu, Rodrigo Lopez-Gonzalez, Nam Chul Kim, Sandra Almeida, et. al.. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 525, 129-133;
- Maria Grazia Biferi, Mathilde Cohen-Tannoudji, Ambra Cappelletto, Benoit Giroux, Marianne Roda, et. al.. (2017). A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Molecular Therapy. 25, 2038-2052;
- Marisa Cappella, Chiara Ciotti, Mathilde Cohen-Tannoudji, Maria Grazia Biferi. (2019). Gene Therapy for ALS—A Perspective. IJMS. 20, 4388;
- Sadanori Miyoshi, Tohru Tezuka, Sumimasa Arimura, Taro Tomono, Takashi Okada, Yuji Yamanashi. (2017).
DOK 7 gene therapy enhances motor activity and life span inALS model mice. EMBO Mol Med. 9, 880-889; - Jan Lejman, Kinga Panuciak, Emilia Nowicka, Angelika Mastalerczyk, Katarzyna Wojciechowska, Monika Lejman. (2023). Gene Therapy in ALS and SMA: Advances, Challenges and Perspectives. IJMS. 24, 1130;
- Cédric Raoul, Toufik Abbas-Terki, Jean-Charles Bensadoun, Sandrine Guillot, Georg Haase, et. al.. (2005). Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 11, 423-428;
- Yuki Hayashi, Kengo Homma, Hidenori Ichijo. (2016). SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Advances in Biological Regulation. 60, 95-104;
- Frank G. (2018). AveXis and its potential ALS gene therapy to be purchased by Novartis for $8.7B. ALS news today;
- Kevin D Foust, Desirée L Salazar, Shibi Likhite, Laura Ferraiuolo, Dara Ditsworth, et. al.. (2013). Therapeutic AAV9-mediated Suppression of Mutant SOD1 Slows Disease Progression and Extends Survival in Models of Inherited ALS. Molecular Therapy. 21, 2148-2159;
- Florie Borel, Gwladys Gernoux, Brynn Cardozo, Jake P. Metterville, Gabriela T. Cabrera, et. al.. (2016). Therapeutic rAAVrh10 MediatedSOD1Silencing in AdultSOD1G93AMice and Nonhuman Primates. Human Gene Therapy. 27, 19-31;
- Christian Mueller, James D. Berry, Diane M. McKenna-Yasek, Gwladys Gernoux, Margaret A. Owegi, et. al.. (2020). SOD1Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N Engl J Med. 383, 151-158;
- Sadanori Miyoshi, Tohru Tezuka, Sumimasa Arimura, Taro Tomono, Takashi Okada, Yuji Yamanashi. (2017).
DOK 7 gene therapy enhances motor activity and life span inALS model mice. EMBO Mol Med. 9, 880-889; - HuiQian Lin, HaoJie Hu, WeiSong Duan, YaLing Liu, GuoJun Tan, et. al.. (2018). Intramuscular Delivery of scAAV9-hIGF1 Prolongs Survival in the hSOD1G93A ALS Mouse Model via Upregulation of D-Amino Acid Oxidase. Mol Neurobiol. 55, 682-695;
- Thomas Gaj, David S. Ojala, Freja K. Ekman, Leah C. Byrne, Prajit Limsirichai, David V. Schaffer. (2017). In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv.. 3;
- Weisong Duan, Moran Guo, Le Yi, Yakun Liu, Zhongyao Li, et. al.. (2020). The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 27, 157-169;
- Pribadi M., Yang Z., Kim T.S., Swartz E.W., Huang A.Y., Chen J.A. et al. (2016). CRISPR-Cas9 targeted deletion of the C9orf72 repeat expansion mutation corrects cellular phenotypes in patient-derived iPS cells. bioRxiv;
- Kai Rothkamm, Stephen Barnard, Jayne Moquet, Michele Ellender, Zohaib Rana, Susanne Burdak-Rothkamm. (2015). DNA damage foci: Meaning and significance. Environ. Mol. Mutagen.. 56, 491-504;
- Rodrigo Lopez-Gonzalez, Dejun Yang, Mochtar Pribadi, Tanya S. Kim, Gopinath Krishnan, et. al.. (2019). Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72 -ALS/FTD. Proc. Natl. Acad. Sci. U.S.A.. 116, 9628-9633;
- Li N. (2022). Strategies for AAV-based therapy of Ducheen muscular dystrophin. Encyclopedia;
- Вылечить миодистрофию Дюшенна: конкуренция групп, единство методик;
- Новое в терапии миодистрофии Дюшенна. (2020). Благотворительный Фонд «Гордей»;
- Na Li, Yafeng Song. (2022). Strategies for Bottlenecks of rAAV-Mediated Expression in Skeletal and Cardiac Muscle of Duchenne Muscular Dystrophy. Genes. 13, 2021.



Комментарии
0Чтобы оставить комментарий, необходимо
войти