Как прочитать эволюцию по генам?
05 ноября 2013
Как прочитать эволюцию по генам?
- 8762
- 0
- 9
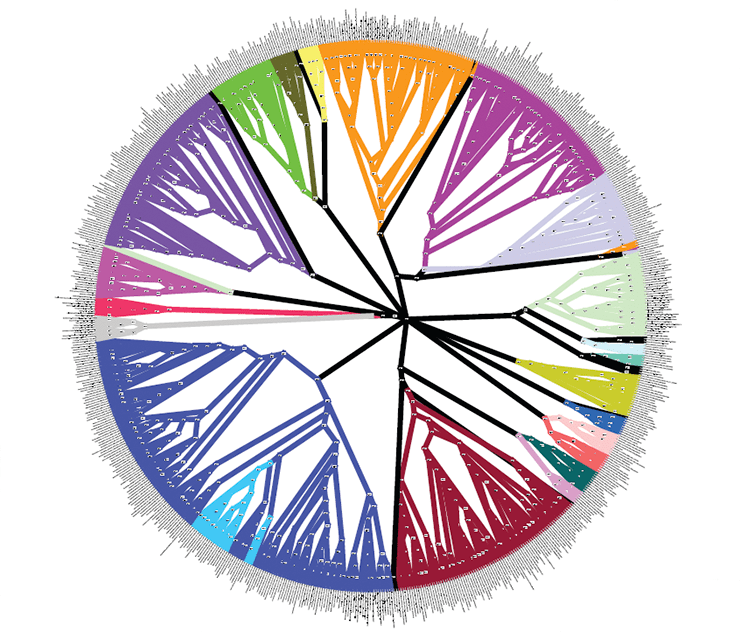
Кладограмма бактерий и архей, построенная на основании 24-х генов с использованием Байесова вывода
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: Проникновение в тайны эволюции — одно из самых захватывающих направлений в современной биологии. Однако тут есть небольшая проблема: пока не изобретена машина времени, чтобы можно было своими глазами увидеть, как развивалась жизнь на Земле. Впрочем, в наше время существуют методики, которые позволяют приподнять завесу тайны над эволюцией, и одна из основных среди них — построение филогении всего живого, то есть «древа жизни». Для этого можно использовать различные признаки, главный среди которых — это последовательность ДНК, в которой закодировано все разнообразие современных и ископаемых существ. В этой статье рассказывается о методиках построения таких филогений, частично заменяющих ученым машину времени.

Конкурс «био/мол/текст»-2013
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2013 в номинации «Лучший обзор».
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
Что такое филогения и филогенетический анализ?
Филогения всех живых существ, или древо жизни, является нашим представлением о степени родства организмов и о том, как шла эволюция живых существ. Кто является ближайшим родственником человека, и каким был наш общий предок? Вымерли ли динозавры, или их потомки до сих пор живут рядом с нами? Произошли ли теплокровность и способность к полету среди позвоночных единожды? Откуда вообще взялись позвоночные? На все эти вопросы уже есть ответы, и получены они были главным образом с помощью филогенетического анализа.
Филогения фактически является той основой, на которую «навешиваются» знания об организмах. Именно она наделяет биологию важным качеством — предсказательностью. Зная те или иные свойства организмов одного вида, с помощью филогении мы можем судить о свойствах родственных ему существ, и даже проследить эволюцию признаков. Древо жизни используется не только в теоретической биологии, но также и в прикладных науках. Например, в медицине и фармакологии филогении используются для того, чтобы понять, откуда были завезены тe или иные вирусы или бактерии, и какие лекарства на них действуют лучше всего [19].
Построение древа жизни является задачей вовсе не тривиальной, и это направление науки, как ни странно, можно считать относительно новым. Разные исследователи пытались проанализировать родственные отношения организмов с самых ранних времен, однако настоящая филогенетическая «революция» случилась только в 50—60-х годах XX века. До 80-х годов деревья строились главным образом на основании морфологических данных, но привлечение ДНК было лишь вопросом времени, поскольку именно в этой молекуле закодированы все признаки организма.
Основные принципы построения филогений
Наверное, самое главное правило, которым руководствуются для построения филогений в наше время — это принцип дихотомии: считается, что из трех таксонов, два должны быть более родственны друг другу, чем третий. Поэтому филогении обычно выглядят как дихотомически разветвленные деревья. Если порядок ветвления установлен для всего дерева, то говорят, что оно полностью разрешенное. Иногда в филогениях бывают «кусты» или политомии — это те места, где порядок ветвления неясен, тогда говорят, что дерево не полностью разрешенное. Этот принцип несовершенен, потому что эволюция таксонов далеко не всегда происходит дихотомически. Когда становится понятно, что дихотомия не отражает реальный случай, исследователи привлекают другие схемы — например, филогенетические сети [8].
Методы построения филогений еще в 60-х годах XX века разделились на две основные ветви — фенетические и кладистические. В то время анализ родственных связей основывался на морфологических признаках [12]; с привлечением к построению филогений молекулярных признаков основные принципы анализа родственных связей остались фактически теми же.
- В фенетике построение филогении основано на общем сходстве двух видов — то есть, чем больше общих признаков, тем ближе они друг к другу;
- В кладистике же считается, что только уникальные для какой-либо группы признаки можно использовать для оценки родства таксонов. Родоначальником кладистического анализа является немецкий ученый Вилли Хенниг [6]. Этот автор также ввел и терминологию, которая широко используется до сих пор. Уникальные признаки называются апоморфиями; ветви, которые объединяются апоморфиями — это клады; а сама филогения называется кладограммой (рис. 2) [12].
Чтобы было более понятно, представьте три вида животных: домашнюю мышь, сумчатую мышь и кенгуру. Домашняя мышь и сумчатая мышь очень похожи друг на друга внешне, но у сумчатой мыши и кенгуру есть общая апоморфия — сумка, — что говорит о том, что эти два вида родственные. Но, естественно, филогенетический анализ основывается на гораздо большем количестве признаков, и группы могут иметь несколько апоморфий.

Рисунок 2. Полностью разрешенная кладограмма. Каждое ветвление — это клада. Обозначенные признаки являются апоморфиями.
Первые шаги. ДНК—ДНК гибридизация
Первые попытки использовать ДНК в качестве основы для построения древа жизни были фенетическими. В 1984 году американские ученые Сибли и Алкист [13] впервые попытались использовать ДНК для прояснения филогении различных видов приматов. Они применили технологию, которая называется «ДНК—ДНК гибридизация». Метод основывается на том, что при копировании в ДНК постоянно происходят мутации. Это приводит к тому, что даже у двух близких родственников последовательности ДНК будут отличаться, не говоря уже о видах. Иными словами, чем дальше находятся организмы на филогенетическом древе, тем больше у них различается ДНК. В данном методе одиночные молекулы ДНК двух видов смешиваются, чтобы они могли образовать «гибридные» двойные спирали, в которых одна половина принадлежит одному виду, а вторая — другому. Затем такие «гибриды» нагреваются, и исследователь смотрит, при какой температуре двойная спираль распадается (или диссоциирует) на две части. Считается, что чем выше температура, требующаяся для распада «гибрида», тем прочнее связь молекул ДНК двух разных видов, и, соответственно, тем ближе эти виды друг к другу (рис. 3).

Рисунок 3. ДНК—ДНК гибридизация. а — Нагревание ДНК двух видов, в результате которого двойная спираль распадается на две части. б — Охлаждение ДНК, в результате которого молекулы ДНК разных видов гибридизуются друг с другом. в — Нагревание ДНК, в результате которого гибридные молекулы ДНК распадаются.
ThinkQuest, рисунок с изменениями
Очень быстро стало понятно, что такой метод не может быть очень точным. Дело в том, что гены могут гибридизоваться не только с гомологичными им генами (гены-ортологи), но и с копиями этих генов, которых в геноме может быть довольно много (гены-паралоги) [15]. Постепенно, с развитием методики секвенирования генов , главным источником для построения филогений стали последовательности ДНК или белков, записанные в виде компьютерных файлов. В последние годы скорость накопления генетической информации растет все увеличивающимися темпами, что окончательно утверждает филогению как метод анализа и обработки биологических текстов.
О стремительном развитии технологий секвенирования ДНК и их роли в науке и обществе можно прочесть в статьях «454-секвенирование (высокопроизводительное пиросеквенирование ДНК)» [20], «Важнейшие методы молекулярной биологии и генной инженерии» [21], «Код жизни: прочесть не значит понять» [22], «Перевалило за тысячу: третья фаза геномики человека» [23] и «Огурцы-убийцы, или как встретились Джим Уотсон и Гордон Мур» [24]. — Ред.
Метод матрицы расстояний (distance matrix)
Метод матрицы расстояний, по сути, является фенетическим. Его основа — расчет попарных различий между соответствующими генами всех видов, участвующих в таком анализе. Делается это следующим образом: гены каждого анализируемого вида сравниваются по каждой позиции нуклеотидов, и чем больше найдено отличий, тем больше будет «расстояние» между видами. Затем строится матрица, в которую заносится это значение для каждой возможной пары сравниваемых генов. Далее матрица расстояний является входной информацией для алгоритмов построения деревьев.
Самый популярный среди подобных алгоритмов — это метод ближайших соседей (neighbour joining). Среди анализируемых видов находят два с минимальными различиями в последовательности (т.е., максимально похожие). Исходя из составленной матрицы, данные об этих видах «объединяются», и далее они участвуют в анализе в объединенном состоянии. Виды один за другим проходят эту процедуру до тех пор, пока не будет найдено одно, полностью разрешенное дерево. Этот алгоритм хорош тем, что он относительно прост и подходит для обработки больших наборов данных (рис. 4) [3].

Рисунок 4. Метод ближайшего соседа
Разные авторы, однако, перечисляют некоторые минусы метода ближайших соседей. Например, есть мнение, что этот метод хуже работает с таксонами, которые филогенетически далеки друг от друга [4], [17]. Также недостатком можно считать и то, что метод всегда выдает дерево с одним-единственным возможным вариантом ветвления [3]. Это происходит потому, что алгоритм подразумевает построение одной филогении без сравнения с другими, тогда как в кладистических методах оцениваются деревья с различным порядком ветвления. Несмотря на то, что в серьезных филогенетических анализах методы матрицы расстояний сейчас почти не используются, они применяются, например, для быстрого построения филогений близкородственных бактерий и вирусов [18].
Метод наибольшей экономии (maximum parsimony)
Этот подход получил большую популярность при анализе морфологических данных, а также какое-то время применялся и для молекулярных исследований. Первый этап анализа — это создание матрицы признаков. Каждый признак должен иметь хотя бы два состояния. Состояний может быть больше, в морфологии они могут описывать разные формы и структуры. Если на кладограмме у какого-то таксона или группы таксонов состояние отличается от предкового, то это называется «переходом из одного состояния в другое». Суть этого алгоритма в том, чтобы найти такое дерево, где присутствует наименьшее суммарное число переходов из одного состояния в другое для всех признаков. В этом случае кладограмма и отображаемая на ней эволюция будут считаться наиболее экономными, а, значит, и более вероятными [3], [12], [16], [17].
Тут возникает вопрос: почему мы вообще считаем, что эволюция должна быть экономной? Дело в том, что это соответствует главному методологическому принципу науки, который заключается в том, что из нескольких равновероятных объяснений надо выбирать наиболее простое, с привлечением как можно меньшего количества сущностей. Этот метод еще называется «Бритвой Оккама». В одной из книг по филогении [3] есть шутливый пример. Представьте, что в одном и том же городе где-то в Северной Америке в соответствующую службу поступает два звонка о том, что по улицам гуляет тигр. Понято, что легче всего предположить, что это один и тот же тигр, который сбежал из зоопарка. Гипотеза, что в городе, где тигров в природе никогда не было, откуда-то появилось сразу же два таких хищника, гораздо менее вероятна.
Эволюция признака — тоже событие нечастое, и когда мы видим два похожих по строению органа, то мы предполагаем, что орган произошел один раз [3]. Это не означает, что признак действительно произошел только один раз, просто это наиболее вероятно. Кладограмма строится на основании многих признаков, и чем больше апоморфий характеризует ту или иную ветвь, тем больше доверия она вызывает.
Плюс метода наибольшей экономии в том, что он интуитивно понятен и довольно прост, но в молекулярных анализах он очень быстро потерял популярность. Один из его недостатков в том, что он не учитывает длину ветвей, которая отображает количество замен нуклеотидов во время эволюции той или иной клады [3]. Некоторые ветви на дереве будут длиннее, потому что скорость эволюции там была выше. При использовании метода наибольшей экономии длинные ветви будут «притягиваться» друг к другу. Этот феномен возникает потому, что чем больше замен нуклеотидов в двух ветвях, тем выше шанс на то, что некоторые из них случайно совпадут, и будут расцениваться как общие апоморфии, даже если это абсолютно не соответствует реальному положению дел.
Другой минус в том, что метод не учитывает разные модели замены нуклеотидов [17]. Например, в методе наибольшей экономии аденин имеет одинаковую вероятность уступить место как тимину, так и цитозину, хотя, как уже отмечалось выше, в организме аденин скорее заменится на цитозин, чем на тимин.
Методы, основанные на моделях эволюции
Наиболее часто используемые методы построения филогений на основе молекулярных данных основываются на моделях эволюции. Один из первых стал метод максимального правдоподобия (maximum likelihood). Для расчета кладограммы, помимо последовательности ДНК, надо выбрать модель замены нуклеотидов, на основании которой будут рассчитываться вероятности. Также в расчет берется длина ветви или эволюционная дистанция между двумя таксонами. Во время анализа рассчитывается, какая длина ветви наиболее вероятна с точки зрения выбранной модели, вероятности всех ветвей кладограммы умножаются, и кладограмма, имеющая наибольшую вероятность, считается правильной [3], [16], [17].
Последний и, наверное, самый популярный в наше время метод — это Байесовский вывод (Bayesian inference). Он, в общем, похож на метод максимального правдоподобия, поскольку также основывается на модели и длине ветвей. Но отличие Байесовского вывода в том, что тут берется в расчет еще один фактор — апостериорная вероятность (posterior probablity), которая рассчитывается на основании как исходных данных, так и полученных результатов анализа [3], [16], [17]. Это не очень понятно интуитивно, но суть в том, что в ходе анализа исследователь получает новые данные, которые тоже можно применить.
Несмотря на все видимые плюсы двух последних методов, тут тоже можно найти некоторые сложности. Главная их слабость в том, что каждый исследователь вынужден подбирать модели самостоятельно, и совсем не обязательно, что он сделает выбор правильно. Но у этой проблемы есть решение. Во-первых, есть программы, которые могут помочь подобрать модель; во-вторых, уже есть алгоритмы на основе Байесовского метода, которые могут «прыгать» с модели на модель, тем самым тестируя их. Еще одна проблема, скорее всего, решаемая с развитием техники, заключается в том, что обсчеты филогений с использованием последних двух методов довольно сложные и требуют много времени и хороших компьютеров.
Все же насколько достоверны филогении?
Думаю, что внимательный читатель заметил, что многие перечисленные методы основаны на вероятностях, и у него может возникнуть закономерный вопрос: как можно доверять филогении, если всегда есть шанс, что построенное дерево ошибочно и не соответствует действительному ходу эволюции? Действительно, методы несовершенны, но на этот вопрос ответ есть.
Во-первых, в филогенетических методах есть понятие «поддержка: чем больше уникальных признаков поддерживают дерево или какую-то его ветвь, тем больше доверия они вызывают [12]. Само дерево может иметь низкую поддержку, зато свидетельств в пользу отдельных его ветвей может быть так много, что корректность не вызовет сомнений. Для подтверждения результата исследователи могут использовать совокупности признаков: последовательности ДНК, РНК и белков, морфологические данные, особенности поведения организмов и многое другое [11]. Когда независимые признаки подтверждают друг друга, уверенность в результате гораздо выше.
Второй ответ на поставленный вопрос еще более обнадеживающий. Его дают эксперименты, проведенные на разных организмах, для которых известна генеалогия, то есть настоящая эволюционная история [1], [5], [7], [10]. Можно привести в пример опыт с мышами, когда филогенетический анализ провели на основе ДНК 24-х линий этих животных. Оказалось, что наблюдаемая последовательность поколений и полученная филогения почти полностью соответствуют друг другу [1]. Это значит, что используемые методы как минимум способны правильно отображать эволюцию.
Плюсы и минусы молекулярных методов построения филогений
У молекулярных методов есть много преимуществ перед морфологическим анализом. Во-первых, ДНК содержит в себе множество данных, которые можно использовать в расчетах, — ведь в генах могут содержаться сотни нуклеотидов. Чаще всего для оценки родства используют больше одного гена, тогда как для анализа на основе морфологических данных используют несколько десятков признаков. Во-вторых, анализ ДНК считается более объективным. Дело в том, что морфологические признаки разные люди могут трактовать и кодировать по-разному, тогда как нуклеотиды всегда одинаковы . В-третьих, ДНК можно использовать как для анализа групп высоких рангов, так и для выяснения отношений между видами, и даже между отдельными индивидами. Морфологический же анализ более достоверен при работе с таксонами высоких рангов, чем на уровне видов, — просто потому, что чем выше ранг, тем лучше отличаются группы, и тем легче отличить аналогичный признак от гомологичного.
В частности, анализ консервативных последовательностей рибосомальных РНК микроорганизмов позволил установить, что все живое на Земле делится не на два царства, как считали несколько десятилетий назад, — эукариот и бактерий, — а на три: эукариот, бактерий и архей. Морфологическое сходство бактерий и архей с лихвой окупается огромной разницей их молекулярного устройства. Честь этого открытия принадлежит Карлу Вёзе. См. также статьи: «Карл Вёзе (1928—2012)», «Эволюция между молотом и наковальней, или как микробиология спасла эволюцию от поглощения молекулярной биологией». — Ред.
Несмотря на то, что преимущество молекулярного анализа кажется вполне обоснованным, есть все же и несколько причин, по которым морфологию нельзя отправить «в отставку».
Первая причина заключается в том, что не каждый организм подходит для выделения ДНК. Он должен быть собран и сохранен специальным образом, иначе эта молекула просто разрушается. Множество редких и интересных видов было описано много десятков лет назад, когда еще даже про ДНК ничего не знали, и в наши дни не очень понятно, где их искать и как собирать. В первую очередь это касается мелких членистоногих, — особенно насекомых, которых чаще всего хранят сухими. То же самое можно сказать и о палеонтологических находках вымерших видов. Для оценки родства таких групп можно использовать только морфологические методы.
Вторая причина заключается в том, что далеко не всегда результаты молекулярных филогенетических методов вызывают доверие. Иногда бывает так, что они не совпадают с устоявшимися «классическими» взглядами. Это, конечно, не означает, что именно молекулярные данные неверны, просто такие несовпадения являются «звоночком», что где-то закралась ошибка. Несовпадения могут быть не только из-за ошибок в самом анализе, но и из-за того, что были неправильно выбраны гены. Гены, мутирующие с высокой скоростью, подходят для выяснения родства между видами, но не походят для анализа групп более высоких рангов. Но гомологичные гены в разных группах организмов могут меняться с разной скоростью, поэтому гены, подходящие для анализа одной группы, могут не подходить для другой группы того же ранга. В общем, подбор нужных участков ДНК может оказаться не очень легкой работой, особенно если учесть, что далеко не все гены у всех видов хорошо изучены.
Третья причина — это высокая стоимость секвенирования генов. Для построения филогении одного небольшого рода можно легко потратить пару тысяч долларов. А если учесть, что гены не всегда подбирают правильно с первого раза, или некоторые экземпляры оказываются непригодными для секвенирования, то анализ надо проводить повторно, и цена может быть больше, чем предполагалось изначально. Анализ же на основе морфологических признаков обходится гораздо дешевле.
Анализ ДНК, безусловно, стал довольно популярным и быстроразвивающимся подходом построения филогений в наши дни. Сейчас специалисты уже используют не просто отдельные гены: в последние годы появились филогенетические исследования на основе более десятка генов или целых митохондриальных геномов [14]. Запущены проекты секвенирования целых геномов разных видов [9], а также проекты для объединения всего живого мира в единое «древо жизни» (рис. 5) [2]. В качестве частного примера можно привести исследование, в результате которого была уточнена «родословная» членистоногих [25]. Наверное, наука сейчас переживает один из самых интересных периодов в развитии анализа ДНК, когда уже видно, что это направление масштабно и многообещающе, и что есть еще очень многое, что нам предстоит узнать о геномах разных организмов. Однако насколько молекулярные методы в филогениях можно развивать, и где граница их применения — покажет будущее.

Рисунок 5. Филогения всех живых существ, или «древо жизни»
Ingmur, рисунок с изменениями
Литература
- W. Atchley, W. Fitch. (1991). Gene trees and the origins of inbred strains of mice. Science. 254, 554-558;
- Assembling Tree of Life (AToL). (2007). The National Science Foundation;
- Baum D.A. and Smith S.D. Tree thinking: an introduction to phylogenetic biology. Greenwood Village, CO: Roberts and Company Publishers, Inc., 2012;
- William J. Bruno, Nicholas D. Socci, Aaron L. Halpern. (2000). Weighted Neighbor Joining: A Likelihood-Based Approach to Distance-Based Phylogeny Reconstruction. Molecular Biology and Evolution. 17, 189-197;
- Douglas T. (1999-2001). 29+ Evidences for Microevolution. TalkOrigins Archive;
- Hennig W. Grundzüge einer Theorie der phylogenetischen Systematik. Berlin: Deutscher Zentralverlag, 1950;
- D. Hillis, J. Bull, M. White, M. Badgett, I. Molineux. (1992). Experimental phylogenetics: generation of a known phylogeny. Science. 255, 589-592;
- V. Kunin. (2005). The net of life: Reconstructing the microbial phylogenetic network. Genome Research. 15, 954-959;
- Jenna Morgan Lang, Aaron E. Darling, Jonathan A. Eisen. (2013). Phylogeny of Bacterial and Archaeal Genomes Using Conserved Genes: Supertrees and Supermatrices. PLoS ONE. 8, e62510;
- Michael M. Miyamoto, Walter M. Fitch. (1995). Testing Species Phylogenies and Phylogenetic Methods with Congruence. Systematic Biology. 44, 64;
- Maureen A. O’Malley, Orkun S. Soyer. (2012). The roles of integration in molecular systems biology. Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences. 43, 58-68;
- Schuh R.T. and Brower A.V.Z. (2003). Biological systematics: principles and applications (2nd Edition). Ithaca: Cornell University Press, 2009;
- Charles G. Sibley, Jon E. Ahlquist. (1984). The phylogeny of the hominoid primates, as indicated by DNA-DNA hybridization. J Mol Evol. 20, 2-15;
- Nan Song, Ai-Ping Liang, Cui-Ping Bu. (2012). A Molecular Phylogeny of Hemiptera Inferred from Mitochondrial Genome Sequences. PLoS ONE. 7, e48778;
- Mark Springer, Carey Krajewski. (1989). DNA Hybridization in Animal Taxonomy: A Critique from First Principles. The Quarterly Review of Biology. 64, 291-318;
- Wiley E.O. and Lieberman B.S. Phylogenetics: the theory of phylogenetic systematics (2nd Edition). Hoboken, NJ: Wiley-Blackwell, 2011;
- Ziheng Yang, Bruce Rannala. (2012). Molecular phylogenetics: principles and practice. Nat Rev Genet. 13, 303-314;
- Longyu Zheng, Tawni L. Crippen, Leslie Holmes, Baneshwar Singh, Meaghan L. Pimsler, et. al.. (2013). Bacteria Mediate Oviposition by the Black Soldier Fly, Hermetia illucens (L.), (Diptera: Stratiomyidae). Sci Rep. 3;
- Мамонты, кости и лекарственная устойчивость: новые технологии позволяют изучать эволюцию возбудителей инфекционных заболеваний;
- 454-секвенирование (высокопроизводительное пиросеквенирование ДНК);
- Важнейшие методы молекулярной биологии и генной инженерии;
- Код жизни: прочесть не значит понять;
- Перевалило за тысячу: третья фаза геномики человека;
- Огурцы-убийцы, или Как встретились Джим Уотсон и Гордон Мур;
- Уточнение «родословной» членистоногих.



Комментарии
0Чтобы оставить комментарий, необходимо
войти