Проблема фолдинга белка
16 октября 2016
Проблема фолдинга белка
- 14413
- 0
- 16
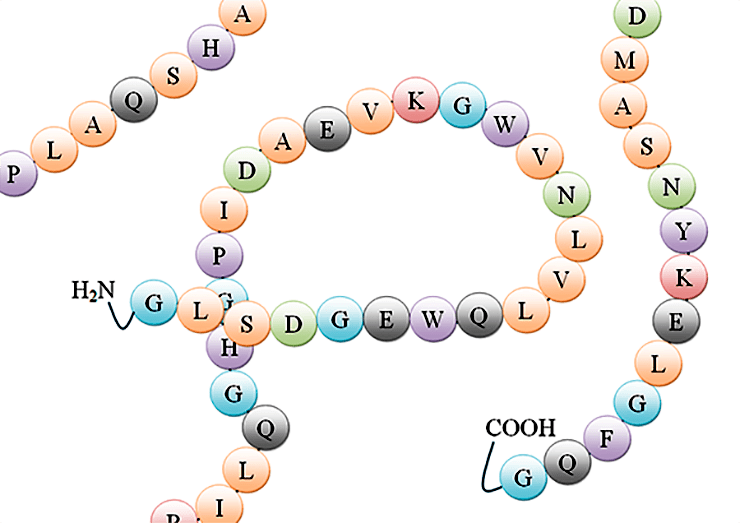
Из такой цепочки аминокислот в результате ее пространственной укладки может получиться хорошо функционирующий белок. Во многом процесс сворачивания белковой цепи пока неясен и представляет одну из крупнейших проблем современной науки.
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: Белки — главные биологические молекулы. Они выполняют множество разнообразных функций: каталитическую, структурную, транспортную, рецепторную и многие другие. Даже всем известная ДНК играет лишь роль «флешки», храня информацию о белках, в то время как белки — сами «файлы». Жизнь на Земле по праву можно назвать белковой. Но так ли много мы знаем о структуре и функционировании этих веществ? До сих пор тайной остается фолдинг белка — процесс пространственной упаковки белковой молекулы, принятия белком строго определенной формы, в которой он выполняет свои функции.

«Био/мол/текст»-2016
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2016.
Генеральным спонсором конкурса, согласно нашему краудфандингу, стал предприниматель Константин Синюшин, за что ему огромный человеческий респект!
Спонсором приза зрительских симпатий выступила фирма «Атлас».
Спонсор публикации этой статьи — Лев Макаров.
Белки — биополимеры, которые можно сравнить с бусами, где бусинами являются аминокислоты, соединенные между собой пептидными связями (отсюда другое название белков — полипептиды). В клетке белки синтезируются на специальных молекулярных машинах — рибосомах. Выходя из рибосомы, полипептидная цепь сворачивается, и белок принимает определенную конформацию, то есть пространственную структуру (рис. 1). Жизненно важно, чтобы белок присутствовал в организме в определенной форме, то есть конформация должна быть «правильной» (нативной). Процесс сворачивания белка и называется фолдингом (от англ. folding — сворачивание, укладка; отметим, что термин «фолдинг» применим не только к белкам). Самое интересное, что информация о трехмерной структуре «заложена» в самой последовательности аминокислот. Таким образом, белку, чтобы принять нативную структуру, требуется лишь «знать», в какой последовательности и какие аминокислотные остатки в нем присутствуют. Впервые это было доказано в 1961 году Кристианом Анфинсеном на примере бычьей панкреатической рибонуклеазы [1] (рис. 2). Следует сказать, что, помимо белков, чья пространственная структура строго определяется аминокислотной последовательностью, существуют так называемые неструктурированные белки (intrinsically unfolded proteins, IDPs): некоторые фрагменты таких молекул, а иногда и целые молекулы, способны принимать сразу множество возможных конформаций, причем все они энергетически «равноценны», а такие белки довольно часто встречаются в природе и выполняют важные функции [2]. Существует и другой тип фолдинга, происходящий с помощью специальных белков — шаперонов, но о нем чуть позже.

Рисунок 1. Котрансляционный фолдинг маленького α-спирального домена. Сворачивание полипептидной цепи многих белков начинается уже в рибосоме во время трансляции белка (то есть его синтеза). Созревающий белок выходит из рибосомы через специальный туннель (на рисунке — затемненная область в большой субъединице), который является важным фактором сворачивания цепи [14], [15], причем С-конец цепи (содержащий карбоксильную группу) фиксирован в рибосоме, а N-конец (содержащий аминогруппу) «продвигается» к выходу и «свисает» из него, когда в туннеле накапливается 30–40 аминокислотных остатков [15]. В туннеле могут формироваться компактизированные незрелые структуры, α-спирали, β-шпильки и маленькие α-спиральные домены [14]. Котрансляционный фолдинг проходит в две стадии: сначала несвернутая цепь (U, unfolded) переходит в компактизированное состояние (C, compacted), которая затем приобретает нативную структуру (N, native).

Рисунок 2. Бычья панкреатическая рибонуклеаза и ученые, которые ее изучали. а — Бычья панкреатическая рибонуклеаза. За исследование структуры этого фермента Анфинсен (Anfinsen) (б), Мур (Moore) (в) и Стайн (Stein) (г) получили Нобелевскую премию по химии (1972 г.) [1], [29]. На примере этого белка впервые было показано явление рефолдинга — самопроизвольного формирования третичной структуры после денатурации (то есть разрушения) [1]. Значение белкового фолдинга заключается в том, что он приводит к формированию строго определенной (нативной) структуры белка, в которой он функционирует. Например, в опыте Анфинсена рибонуклеаза в результате рефолдинга восстановила свою ферментативную активность, то есть стала вновь хорошо катализировать биохимическую реакцию. Для того чтобы этот фермент работал, в единый каталитический центр (один кусочек пространства) должны из совершенно разных мест белковой цепи собраться пять аминокислотных остатков: гистидин (12), лизин (41), треонин (47), гистидин (119) и фенилаланин (120) [21].
модель из базы данных PDB (PDB ID 5D6U), портреты ученых с сайта ru.wikipedia.org
Актуальность проблемы
Проблема заключается в том, что человечество со всеми своими вычислительными мощностями и арсеналом экспериментальных данных до сих пор не научилось строить модели, которые бы описывали процесс белкового фолдинга и предсказывать трехмерную структуру белка на основе его первичной структуры (то есть аминокислотной последовательности). Таким образом, полного понимания этого физического процесса до сих пор нет.
Взрывной рост геномных проектов привел к тому, что секвенируется все больше геномов, а соответствующие последовательности ДНК и РНК наполняют базы данных по экспоненте. На рис. 3 изображены рост числа аминокислотных последовательностей, а также рост числа известных белковых структур в период с 1996 по 2007 годы. Хорошо видно, что число известных структур значительно меньше, чем число последовательностей. На момент написания настоящей статьи (август 2016 г.) число последовательностей в базе данных UniParc составляет более 124 миллионов, в то время как количество структур в базе данных PDB (Protein Data Bank) — лишь чуть больше 121 тысячи, что составляет менее 0,1% от всех известных последовательностей, причем разрыв между двумя этими показателями стремительно нарастает и, вероятно, будет расти и дальше. Такое сильное отставание связано с относительной сложностью современных методов определения структур [3]. При этом знать их очень важно. Поэтому вопрос применения вычислительных методов с целью предсказания белковых структур по их последовательностям стоит сейчас остро. В 2005 году авторитетный журнал Science признал проблему фолдинга белка одной из 125 крупнейших проблем современной науки [4].

Рисунок 3. Сравнение темпов роста числа известных последовательностей и структур с 1996 по 2007 годы. На горизонтальной оси указываются годы, на левой вертикальной — число последовательностей в миллионах (сплошная линия), на правой вертикальной — число структур в миллионах (пунктирная линия). Четко видно отставание количества известных структур от количества последовательностей. К настоящему моменту разрыв вырос еще сильнее.
После прочтения генома человека стали известны многие человеческие гены и, следовательно, аминокислотные последовательности, кодируемые ими [5]. Однако это не значит, что мы знаем функции всех генов, иначе говоря, мы не знаем функции белков, кодируемых этими генами. Известно, что во многом функции белков можно предсказать по их структуре, хоть и не всегда [6], [7]. Поэтому заветной мечтой является способность предсказывать структуру и, как следствие, функцию белка по самой нуклеотидной последовательности гена.
Что делается для решения проблемы?
Неверно, однако, думать, что мы не знаем совсем ничего. Конечно, накоплено большое количество фактов о фолдинге, известны закономерности этого процесса, разработаны различные методы его моделирования [3], [6–8]. Чтобы отслеживать успехи, достигаемые на пути к решению проблемы фолдинга, был создан международный конкурс по предсказанию пространственной структуры белковых молекул — CASP (Critical Asessement of techniques for protein Structure Prediction), проходящий раз в два года (сейчас соревнование проходит в двенадцатый раз, оно началось в апреле и закончится в декабре 2016 года). В этом состязании исследователи соревнуются, кто лучше предскажет структуру белка по его аминокислотной последовательности, причем конкурс проходит с использованием двойного слепого метода (на момент проведения конкурса структура белка-«загадки» попросту неизвестна; ее определение завершается каждый раз по окончании состязания). Пока что структуры белков-мишеней точно не были предсказаны ни разу.
Существует две группы методов предсказания структуры.
К первой относятся так называемые методы моделирования «с нуля» (ab initio, de novo, есть и другие синонимичные термины), когда модели строятся лишь на основании первичной структуры, без использования сравнительных методов с уже известными структурами, зато с использованием всего накопленного понимания физики сворачивания биополимеров. Фундаментальная значимость этих методов состоит в том, что они помогают понять физико-химические принципы белкового фолдинга, ответить на этот животрепещущий вопрос — почему белок сворачивается так, а не иначе? Однако недостатками этих методов являются очень большая сложность вычисления и невысокая точность [9]. Эти методы требуют упрощений и приближений, а также являются неэффективными для предсказания структур крупных белков. В 2007 году за счет методов моделирования de novo впервые с высокой точностью была определена структура одного из белков бактерии Bacillus halodurans, но белок этот относительно невелик (112 аминокислотных остатков), а для получения точной модели потребовалась мощность более 70 000 персональных компьютеров и суперкомпьютера; кроме того, из 26 полученных моделей точной оказалась лишь одна [10]. Методы молекулярной динамики (МД) позволяют описывать молекулярные события [11] и способны проследить процесс сворачивания белка в нативную структуру: в 2010 году впервые удалось это сделать за счет вычислительной мощности специально созданного суперкомпьютера Anton [12].
Ко второй группе методов относятся методы сопоставительного моделирования. Они основываются на явлении гомологии, то есть общности происхождения объектов (органов, молекул и др.). Таким образом, у «предсказателя» есть возможность сравнивать последовательность белка, структуру которого необходимо смоделировать, с шаблоном, то есть белком, структура которого известна и который предположительно является гомологом, и на основании их схожести строить модель с последующими корректировками (похожие последовательности сворачиваются в похожие структуры). Эти методы сейчас более популярны, так как предсказание структуры белков является важной практической задачей, а к настоящему моменту появились вычислительные средства, базы данных, а также стало известно, что количество возможных вариантов укладок белковых структур ограничено [6], [9] (рис. 4). И пусть эти методы не снимают самой проблемы белкового фолдинга, они способны помочь решать конкретные практические задачи, пока другие бьются над исследованием более фундаментальных вопросов.

Рисунок 4. Динамика выявления новых типов фолдов (вариантов упаковки). На горизонтальной оси откладывается время (годы), на левой вертикальной оси — доля новых фолдов (более детально — на вкладке) (сплошная линия), а на правой вертикальной оси — общее число структур (пунктирная линия), классифицированных в базе данных CATH. Отметим, что эта база данных занимается структурной классификацией белков, поэтому для нее принципиально знать возможные типы белковых фолдов. Явно видно, что со временем классифицируется все больше и больше белков, но при этом количество вариантов фолдов уменьшается.
Нужно подчеркнуть, что современные методы предсказания белковых структур требуют большой вычислительной мощности и часто осуществляются на суперкомпьютерах или с помощью сетей распределенных вычислений , как, например, Rosetta@home и Folding@home. К участию в работе этих проектов приглашаются все желающие: нужно лишь запустить программу на своем компьютере, пока он не нужен пользователю.
Подробнее о применении компьютерных методов для предсказания белковых структур читайте в статье на «биомолекуле»: «Торжество компьютерных методов: предсказание строения белков» [3].
Некоторые закономерности фолдинга белка
Известны некоторые закономерности белкового фолдинга. Сейчас считается, что этот процесс происходит поэтапно: сначала линейная цепочка, имеющая нулевую энтропию, быстро сворачивается с образованием статистического клубка — энтропийное сворачивание [13]. Затем происходит гидрофобный коллапс: гидрофобные аминокислотные остатки «прячутся» вглубь молекулы, а гидрофильные — «расселяются» по поверхности (см. ниже). Результатом этого этапа является формирование расплавленной глобулы. После этого происходит формирование специфических связей (см. ниже), и белок переходит в состояние истинной глобулы, при этом свободная энергия резко падает .
Последний этап не происходит при фолдинге неструктурированных белков — IDPs.
Нужно отметить, что для каждой аминокислотной последовательности теоретически можно предположить множество путей, которыми она может идти для достижения нативной конформации. Однако известно, что белок не перебирает все возможные варианты, а движется по одному из возможных путей, определенных для каждой последовательности. Если бы белок пробовал все возможные варианты, то время пути от простой последовательности к нативному состоянию превысило бы время существования Вселенной (парадокс Левинталя)! Конечно, такого не происходит: время принятия белком нативной структуры составляет доли секунды. Это похоже на сборку кубика Рубика: из состояния несобранного кубика к состоянию собранного можно прийти множеством разнообразных путей, однако на соревнованиях по скорости сборки кубика побеждает тот, кто делает это быстрее и эффективнее, то есть выбирает определенный путь. На самом деле найти такой путь — и есть главная задача методов моделирования ab initio (см. выше). Ответ на фундаментальный вопрос фолдинга будет заключаться не просто в способности безошибочно моделировать структуры, а, в первую очередь, в том, чтобы знать и обосновывать путь достижения белком нативного состояния.
Следует подчеркнуть значение котрансляционного фолдинга (рис. 1), о котором говорилось выше, в формировании структуры белка. Отметим, что присутствие рибосомы, на которой синтезируется белок, накладывает серьезные коррективы на процесс сворачивания цепочки. Это всегда нужно иметь в виду при моделировании фолдинга природных белков in vivo. Канал, в котором оказывается растущая цепь, ограничивает ее конформационную изменчивость, а потому далеко не все типы структур могут в ней формироваться [14], [15]. Кроме того, растущая цепочка постоянно проталкивается вперед (на один аминокислотный остаток при каждом акте транспептидации-транслокации, то есть образования новой пептидной связи и последующего продвижения рибосомы), а потому логично будет предположить, что конформация цепи в рибосомном канале обладает такими качествами, как жесткость и векторность, что соответствует свойствам α-спирали [15]. Кроме того, взаимная ориентация аминокислотных остатков в двух центрах внутри рибосомы всегда однотипная (эквивалентная), не зависящая от природы этих остатков, что тоже, по-видимому, способствует формированию α-спиралей [15]. Действительно, α-спирали — наиболее типичный элемент вторичной структуры белков. Они были открыты Лайнусом Полингом (Liunus Pauling) и Робертом Кори (Robert Corey), которые вместе с Уолтером Колтуном (Walter Koltun) предложили новый тип моделей молекул [16].
В то же время, когда N-конец (содержащий аминогруппу) растущей цепи белка выходит из туннеля и погружается в раствор, на него начинают действовать физико-химические условия этой среды, и белок начинает подчиняться их правилам.
Известный молекулярный биолог академик Александр Спирин [15] в этой связи отмечает три различия между фолдингом in vitro и in vivo:
- Во-первых, различна стартовая конформация: если в экспериментальных условиях ренатурация начинается с некоего состояния развернутой цепочки в растворе, то в случае с рибосомой фолдинг начинается уже с какой-то определенной конформации, обеспеченной рибосомальным каналом.
- Во-вторых, при котрансляционном фолдинге сворачивание начинается с N-конца, то есть процесс фолдинга направленный, а в случае фолдинга без участия рибосомы поиск конформаций осуществляется сразу всей молекулой.
- Третье отличие заключается в том, что в случае котрансляционного фолдинга C-конец белковой цепи фиксирован рибосомой, относительно крупной частицей, что приводит к стабилизации промежуточных структур (см. выше), а в случае рефолдинга in vitro такой стабилизации не происходит.
Эти соображения лишний раз доказывают, что биологические вопросы не могут решаться «всухую» за счет применения методов биоинформатики [17]. Даже самые, казалось бы, выверенные компьютерные модели могут оказаться неточны, если они построены без учета факторов, реально действующих в природе.
«Сухая» биология — та, которая использует компьютерные и математические методы, в то время как «мокрая» — это работа с растворами, пипетками, микроскопами и т. д.: «Вычислительное будущее биологии» [18] и «Я б в биоинформатики пошел, пусть меня научат!» [19].
Для решения проблемы фолдинга разработаны так называемые эмпирические потенциалы: парных взаимодействий остатков, водородных связей, торсионных углов, центров масс боковых цепей и многие другие [6], [11]. Например, потенциал сольватации позволяет предсказать, внутри или снаружи белка будет находиться аминокислотный остаток (соответственно заглубленный или экспонированный) в зависимости от его гидрофобности [6], [13]. Известно, что одни аминокислоты «любят» воду (гидрофильные), они будут с большей вероятностью располагаться на поверхности белковой молекулы, а другие — «не любят» (гидрофобные) и «прячутся» в более недоступные для растворителя области молекулы, заслоняясь другими остатками (рис. 5). Гидрофобный эффект имеет большое значение в фолдинге белка.

Рисунок 5. Гидрофобность аминокислот влияет на их пространственное распределение (на примере одной из человеческих дегидрогеназ). Гидрофильные аминокислоты показаны синим цветом, гидрофобные — красным. Можно заметить, что гидрофильные остатки стремятся располагаться на открытых для растворителя участках, в то время как гидрофобные — в закрытых областях молекулы.
база данных PDB (PDB ID 5ICS)
Важным аспектом формирования структуры белка на всех этапах является образование связей между радикалами (боковыми цепями) аминокислотных остатков. Они бывают разные: гидрофобные, электростатические и другие [20]. Интересным вариантом является формирование дисульфидных связей («мостиков») за счет взаимодействия атомов серы боковых цепей цистеина. Например, в прославленной рибонуклеазе, за исследование структуры которой была дана Нобелевская премия, таких связей четыре. Однако здесь все не так просто. Если в состав белковой цепи входят два атома серы, принадлежащие цистеину, то легко сказать, что может образоваться один дисульфидный мостик. Но если атомов серы, к примеру, десять и, соответственно, образуются пять SS-связей, то мы не можем однозначно сказать, какие именно атомы серы будут попарно взаимодействовать друг с другом (а белок может). Согласно расчетам Томаса Крейтона (Thomas Creighton), если в белке 5 дисульфидных связей, число возможных комбинаций составляет уже 945, если таких связей 10, то число вариантов составляет 654 729 075, а при 25 дисульфидных связях это число превышает 5 квадриллионов квадриллионов (более 5,8 × 1030) [21]. А в белке реализуется лишь один вариант, и притом всегда один и тот же! Следует тем не менее отметить, что это справедливо для самоорганизации белков in vitro («в пробирке», «в стекле», то есть в условиях эксперимента, а не в живом организме) в подходящих условиях, а in vivo (в живом организме) самоорганизации дисульфидных связей не происходит. Их образование катализирует специальный фермент — протеиндисульфидизомераза [7], [22] или ПДИ, которая к тому же способна «исправлять» ошибки в случае неправильного образования SS-связи, таким образом корректируя процесс фолдинга [22], [23].
Важно понимать, что процесс формирования окончательной структуры белка не заключается лишь в простом сворачивании цепочки. В клетках белки подвергаются ацетилированию, гликозилированию и многим другим модификациям. Поэтому, например, количество разных аминокислот в белках превышает известные 20 («магическая двадцатка», по образному выражению нобелевского лауреата Фрэнсиса Крика). Кроме того, для формирования сложных (олигомерных) белков необходимо формирование специфических связей между отдельными протомерами (например, в молекуле гемоглобина четыре протомера, то есть отдельно синтезированные цепочки). Для многих белков, особенно ферментов, важным является присоединение простетической группы, то есть небелкового компонента. Могут происходить и другие преобразования [7].
Известны многие другие закономерности белкового фолдинга. Завеса тайны постепенно приподнимается. Однако картина до сих пор далека от целостной. Успехи предсказания структур пока только эпизодические. В связи с этим научное сообщество сделало следующий любопытный шаг: оно привлекло к решению вопроса широкую общественность, создав игру FoldIt [24], [25]. Принять участие в мировом соревновании может любой желающий. Суть игры заключается в том, чтобы свернуть белковую цепочку максимально компактно, то есть привести белковую молекулу в такое состояние, в котором свободного места внутри клубочка как можно меньше — именно в таком виде белки присутствуют в природе (рис. 6). С точки зрения термодинамики, такому состоянию соответствует минимум свободной энергии [7], [26]. Чем более компактная молекула получается, чем меньше полостей и открытых гидрофобных участков, чем больше открытых гидрофильных участков, водородных связей в структурах типа β-листов, чем меньше «столкновений» атомов, тем большее количество баллов игроку начисляется. Таким образом, наибольшее количество баллов получает модель с наименьшей свободной энергией. Большинство игроков FoldIt имеют лишь малую биохимическую подготовку либо не имеют ее вовсе [27]. Игра основана на алгоритмах Rosetta и не является моделированием структур de novo, которое, как верно подмечают авторы, все еще остается исключительно сложной проблемой [27].

Рисунок 6. Сравнение разных форм представления моделей белковых структур (на примере одной из человеческих трансфераз). а — Форма, наглядно демонстрирующая типы вторичных структур. б — Форма, показывающая реальное расположение атомов молекулы белка в пространстве (Space Fill). Хорошо видно, что молекулы белков сильно компактизированы, между атомами мало свободного пространства.
база данных PDB (PDB ID 5CU6)
Группа игроков FoldIt принимает участие в CASP. Игра уже показала свою эффективность в предсказании структур и даже бóльшую эффективность в сравнении с другими методами, а также решила серьезную научную проблему структуры протеазы вируса иммунодефицита обезьян, которую наука не могла решить на протяжении более чем десятилетия [27].
Говоря о применении разных методов и средств для решения обсуждаемой проблемы, всегда нужно помнить, что не все последовательности могут сворачиваться строго определенным образом. Вероятно, мы, глядя на результаты, к которым пришла эволюция к настоящему времени, видим только те последовательности, которые могут сворачиваться, поскольку они хорошо выполняли свои функции и были поддержаны отбором.
«Гувернантки» для белков — шапероны
Говоря о фолдинге, мы акцентировали внимание на относительной автономности этого процесса: белковая молекула принимает определенную конформацию на основании своей первичной структуры, и происходит это в конкретных (что важно) физико-химических условиях (кислотность, температура, природа растворителя и др.). Тем не менее не должно складываться впечатление, будто бы фолдинг абсолютно независим, особенно для крупных белков. Мы лишь упомянули о ферменте ПДИ, помогающем белку правильно свернуться. Кроме этого фермента, есть и другие (например, ППИ — пептидил-пролил-цис/транс-изомераза [22], [23]). Но ферменты — не единственная группа белков, помогающая правильно сворачиваться другим белкам. Существует еще одна особая группа белков, играющих важную роль в фолдинге. Называются они шаперонами.
Шапероны — сложные белки с консервативным (то есть эволюционно малоизменчивым) механизмом действия, встречающиеся во всех царствах живой природы. Это и понятно: их роль в жизнедеятельности клетки огромна . Как говорилось выше, созревающая белковая цепочка выходит из рибосомы. Она еще незрелая, а пребывает в так называемом «расплавленном» состоянии. Такие незрелые молекулы подвержены дурному влиянию окружения: они могут взаимодействовать с другими клеточными белками, образуя агрегаты, что может приводить к болезням, например, болезни Альцгеймера или Паркинсона. Но есть и «правильное» русло, по которому может (и должно) быть направлено развитие белка, — тот путь, который приведет расплавленную глобулу в нативное состояние. Тут и помогают шапероны, «подкарауливая» и захватывая белковые цепочки у самого выхода из рибосомного туннеля и таким образом направляя незрелые белки, находящиеся на судьбоносном перепутье, в верное русло. Шапероны названы так неспроста: раньше в Англии так называли пожилую опытную даму, которая сопровождала молодую девушку, впервые вышедшую в свет под ее руководством, и удерживала ее от непродуманных контактов [21]. (Термин «шаперон» и сейчас используется в близких значениях.) Шапероны не являются специфичными для разных аминокислотных последовательностей зарождающихся цепей, но они могут отличать зрелые белки от незрелых и действуют на последних.
На «биомолекуле» уже была статья, посвященная шаперонам: «Канал эукариотического шаперонина открывается подобно диафрагме фотоаппарата» [28].
Важнейшая группа шаперонов — шаперонины. Интересна их структура: они представляют собой бочонки, составленные из двух колец. Сворачивающийся белок попадает внутрь шаперонина, а «вход» закрывается специальной «шапочкой» либо смыканием краев блоков, из которых состоят кольца [28], чтобы белковая молекула не покинула шаперонин раньше времени (рис. 7). В таком защищенном состоянии белок может окончательно принять нативную конформацию. Пока малопонятны процессы, происходящие внутри бочонков-шаперонинов.

Рисунок 7. Схематическое изображение двух типов шаперонинов — I и II. а — Шаперонины I типа характерны для бактерий (шаперон GroEL имеет структуру бочонка, составленного из двух колец, в каждом — 7 «блоков»; внутри шаперонина — камера, в которой происходит превращение расплавленной глобулы в нативную; бочонок закрывается «крышкой» — GroES); б — Шаперонины II типа, характерные для архей и эукариот (здесь каждое из двух колец состоит из 8 «блоков»; закрытие камеры происходит не за счет присоединения «крышки», а по механизму объектива фотоаппарата [28]).
Нужно сказать, что шапероны не только участвуют в фолдинге созревающих цепей, но и помогают «сломанным» белковым структурам, которые возникли в клетке в результате определенных воздействий, вновь принять правильную конформацию. Наиболее типичная причина таких «поломок» — тепловой шок, то есть поднятие температуры. В связи с этим часто употребляют другие названия шаперонов — белки теплового шока (heat shock proteins, hsp) или белки стресса. Шапероны выполняют другие важные функции в клетке, например, транспорт белков через мембраны и сборку олигомерных белков.
Заключение
Итак, для фолдинга белка строго необходимы следующие условия: первичная структура, конкретные физико-химические условия, а также две группы вспомогательных белков — специфически работающие ферменты и неспецифически работающие шапероны.
Резюмируя, скажем, что белковый фолдинг — одна из центральных проблем современной биофизики. И хотя накоплен большой арсенал данных об этом явлении, до сих пор оно малопонятно, что выражается, в конечном счете, в невозможности предсказания трехмерной структуры на основании аминокислотной последовательности (особенно это касается крупных, в том числе олигомерных, белков). Успехи в этой области, а особенно моделирования de novo, пока можно назвать единичными. Тем не менее понятно, что решение точно есть, ведь в природе белки почти всегда сворачиваются одинаковым, правильным образом. Существует как фундаментальная, так и большая практическая значимость «взлома» кода белкового фолдинга. А это значит, что работы в этой области будут вестись и дальше.
Литература
- Anfinsen C.B., Haber E., Sela M., White F.H. Jr. (1961). The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. 9, 1309–1314;
- За пределами порядка;
- Торжество компьютерных методов: предсказание строения белков;
- So much more to know. (2005). Science. 309, 78–102;
- Геном человека: как это было и как это будет;
- Rigden D.J. From protein structure to functions with bioinformatics. Springer Science + Business Media B.V., 2009. — 328 p.;
- Финкельштейн А.В. и Птицын О.Б. Физика белка: Курс лекций с цветными и стереоскопическими иллюстрациями и задачами (3-е изд., испр. и доп.). М.: КДУ, 2012. — 456 с.;
- Иванов В.А., Рабинович А.Л., Хохлов А.Р. Методы компьютерного моделирования для исследования полимеров и биополимеров. М.: Либроком, 2009. — 662 с.;
- Greene L.H., Lewis T.E., Addou S., Cuff A., Dallman T., Dibley M. et al. (2007). The CATH domain structure database: new protocols and classification levels give a more comphensive resource for expoling evolution. Nucleic Acids Res. 35, D291—D297;
- Новые успехи в предсказании пространственной структуры белков;
- Молекулярная динамика биомолекул. Часть I. История полувековой давности;
- Миллисекундный барьер взят!;
- Физическая водобоязнь;
- Rodnina M.V. and Wintermeyer W. (2016). Protein elongation, co-translational folding and targeting. J. Mol. Biol. 10, 2165–2185;
- Спирин А.С. Молекулярная биология: структура рибосомы и биосинтез белка. М.: Высшая школа, 1986. — 303 с.;
- На заре молекулярной графики;
- Penders B., Horstman K., Vos R. (2007). Proper science in moist biology. EMBO Rep. 7, 613;
- Вычислительное будущее биологии;
- Я б в биоинформатики пошёл, пусть меня научат!;
- Роль слабых взаимодействий в биополимерах;
- Филиппович Ю.Б. Основы биохимии: Учебник для студентов высших учебных заведений, обучающихся по направлению и специальности «Химия» и «Биология» (4-е изд., перераб. и доп.). М., С.-П.: Агар, Флинта, Лань, 1999. — 512 с.;
- Наградова Н.К. (1996). Внутриклеточная регуляция формирования нативной пространственной структуры белков. Соросовский журнал. 7, 10–18;
- Эллиот В. и Эллиот Д. Биохимия и молекулярная биология / Под ред. А.И. Арчакова, М.П.Кирпичникова, А.Е.Медведева, В.П. Скулечева; Пер с англ. О.В. Добрыниной, И.С. Севериной, Е.Д. Скоцеляс и др. М.: МАИК «Наука/Интерпериодика», 2002. — 446 с.;
- Элементы: «Помогать науке можно играя»;
- Тетрис XXI века;
- Недоупорядоченные белки;
- Khatib F., DiMaio F., Foldit Contenders Group, Foldit Void Crushers Group, Cooper S., Kazmierczyk M. et al. (2011) Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat. Struct. Mol. Biol. 18, 1175–1177;
- Канал эукариотического шаперонина открывается подобно диафрагме фотоаппарата;
- Anfinsen C.B. (1973). Principles that govern the folding of protein chains. Science. 181, 223–230.



Комментарии
0Чтобы оставить комментарий, необходимо
войти