Муковисцидоз — первые надежды
31 октября 2018
Муковисцидоз — первые надежды
- 20277
- 0
- 8
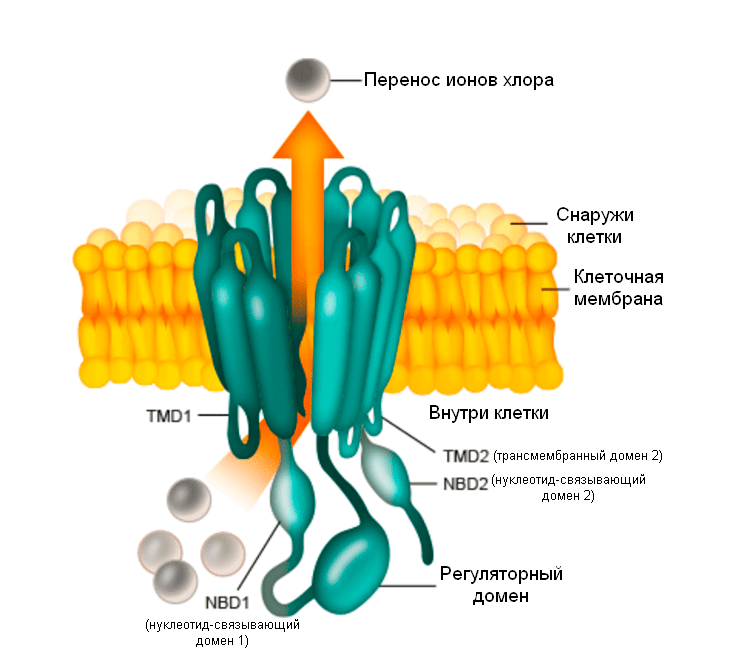
CFTR — хлорный канал, мутации в котором являются причиной муковисцидоза. TMD — трансмембранный домен.
[1], рисунок адаптирован
-
Автор
-
Редакторы
Темы
Статья на конкурс «био/мол/текст»: Муковисцидоз — самое распространенное из моногенетических заболеваний (обусловленных поломкой одного гена). При нем нарушено функционирование белка-переносчика ионов хлора через мембрану клетки — хлорного канала CFTR. Так как этот канал отвечает за нормальную работу эпителия в легких, кишечнике, поджелудочной железе и других органах, его дисфункция приводит к накоплению в этих органах слизи, повышению вероятности инфекций и в конце концов к преждевременной смерти. До последнего времени врачи могли лечить только симптомы муковисцидоза: разжижать слизь, расширять бронхи, снижать воспаление, а также уничтожать бактерий антибиотиками, причем все эти меры почти не продлевали жизнь. Но за последние годы был достигнут невиданный прогресс: средняя продолжительность жизни больных возросла более чем в два раза. В этой статье будет рассказано о препаратах, благодаря которым стал возможен такой успех, об истории их создания и перспективах. На данных примерах читатель также узнает, как происходит современная разработка лекарств.

Конкурс «био/мол/текст»-2018
Эта работа опубликована в номинации «Биофармацевтика» конкурса «био/мол/текст»-2018.
Партнер номинации — медицинская компания «Инвитро».
Генеральный спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Спонсором приза зрительских симпатий выступил медико-генетический центр Genotek.
«Книжный» спонсор конкурса — «Альпина нон-фикшн»
Описание заболевания
Вначале рассмотрим подробнее, что за болезнь муковисцидоз и почему разработка лекарств против него оказалась таким непростым делом.
Большинство случаев этого страшного заболевания диагностируют в первые годы жизни, потому что муковисцидоз поражает все органы, но особенно — легкие и кишечник. Больные страдают от многочисленных нарушений работы почти всех систем организма: дыхательной, пищеварительной, опорно-двигательной, нервной, сердечно-сосудистой и других. Средняя продолжительность жизни больных составляет 30–40 лет (и сильно зависит от качества ухода), 90% пациентов умирает от легочных осложнений.
Муковисцидоз (или кистозный фиброз, как он называется по-английски) возникает у тех людей, у которых плохо работает или отсутствует белок CFTR. Его название расшифровывается как cystic fibrosis transmembrane conductance regulator, то есть регулятор трансмембранной проводимости при муковисцидозе. Сейчас разберемся, какова роль CFTR в норме и почему его недостаток приводит к таким тяжелым последствиям.
CFTR относится к трансмембранным белкам , которые связывают АТФ и меняют за счет этого свою конформацию. Внутри белка открывается канал, который позволяет ионам хлора выходить из клетки наружу. После гидролиза АТФ канал закрывается (рис. 1).
То есть он пронизывает мембрану клетки насквозь.
АТФ — аденозинтрифосфорная кислота, основная молекула, которая запасает и передает химическую энергию в клетках.

Рисунок 1. Диаграмма предполагаемой структуры белка CFTR в закрытом (слева) и открытом (справа) положениях. Два трансмембранных домена образуют канал. Открытие канала контролируется двумя внутриклеточными доменами (NBD1 и NBD2), которые способны связывать и гидролизовать АТФ (голубой). Регуляторный домен (зеленый) содержит сайты фосфорилирования (Р). Активация канала требует наличия остатка фосфорной кислоты на регуляторном домене. NBD1 и NBD2 связывают и гидролизуют АТФ, вызывая открытие канала путем взаимодействия с трансмембранными доменами. Одна из молекул АТФ остается связанной с NBD1 в течение нескольких минут. За это время происходит несколько циклов открывания-закрывания канала, обусловленных связыванием и гидролизом второй молекулы АТФ [4], [5].
Наличие ионов вблизи поверхности клеток необходимо для поддержания нормального осмотического давления, а это важно для циркуляции жидкости в околоклеточном пространстве. Поэтому постоянный контролируемый поток ионов хлора через мембрану необходим для нормального функционирования эпителия легких, кишечника, протоков поджелудочной железы, яичников, потовых желез.
При муковисцидозе в первую очередь поражаются именно эти органы: в железах образуется густая слизь, которая забивает протоки и мешает нормальной работе органов. А вот почему в легких и кишечнике нарушается работа врожденной иммунной системы, возникает хроническое воспаление и инфекции — стало более-менее понятно совсем недавно. На нынешний момент картина примерно такая: снижение концентрации ионов хлора в околоклеточном пространстве вызывает активацию эпителиального натриевого канала ENaC, который начинает закачивать натрий в клетку. Уменьшение концентрации NaCl возле поверхности клетки вызывает снижение осмотической силы, а, следовательно, количества воды, поступающей к клетке. В случае легких это приводит к осушению воздушных путей и снижению очищающей активности ресничек и слизистой оболочки (рис. 2 во врезке).
Постоянный ток слизи вдоль поверхности воздушных путей очень важен для функционирования защитной системы легких. При этом слизь должна быть достаточно жидкой, чтобы растекаться по поверхности эпителия после секреции, но и достаточно вязкой, чтобы движение ресничек эпителия вызывало ее направленный ток. У больных муковисцидозом слизь содержит слишком мало воды, поэтому она густая и неподвижная, от нее трудно избавиться даже при кашле. Собственно, русское название болезни и происходит от двух латинских слов: mucus («слизь») и viscosus («вязкий»).
Поскольку слизь — это полимерная сетка, образованная белками, она содержит поры. Характерная черта слизи при патологии — слишком мелкий размер пор. В нормальной слизи они имеют диаметр 0,2–1 мкм, а при болезни — менее 0,1 мкм. В результате нейтрофилы — клетки иммунной системы, которые в первую очередь отвечают за защиту от бактерий, — не могут проникнуть изнутри сквозь слой слизи. Бактерии на поверхности воздушных путей размножаются беспрепятственно и вызывают хронические инфекции, которые являются главной причиной смертности при муковисцидозе (80% пациентов) [7]. Более того, на уплотненной слизи, в отличие от обычной, бактерии образуют макроколонии, так называемые биопленки, которые особенно устойчивы к действию иммунной системы и антибиотиков [8].
Из сказанного понятно, что одним из средств улучшить состояние больных муковисцидозом должна быть регидратация легких, которую можно обеспечить с помощью ингаляции гипертонического раствора соли. Это паллиативная мера, не воздействующая на причину болезни, но, тем не менее, она позволяет снизить количество осложнений [6].
До открытия молекулярных причин болезни пациентов лечили только симптоматически — разжижая слизь, применяя антибактериальные, противовоспалительные препараты и физиотерапию. У пациентов с нарушениями ЖКТ и поджелудочной железы эффективна терапия диетами и пищеварительными ферментами. Все эти меры облегчают состояние пациентов, однако настоящий скачок в продлении жизни и улучшении ее качества стал возможным только благодаря открытиям молекулярной биологии и рациональной разработке лекарств [9], [10].
Фокус на CFTR
В 1989 году был найден ген CFTR , кодирующий хлорный канал длиной 1480 аминокислот, и начат поиск мутаций, отвечающих за развитие муковисцидоза. Всего описано более 2000 мутаций в гене CFTR, однако только 250–300 из них приводят к муковисцидозу, а достаточно часто встречается (более чем у 0,1% больных) примерно 20 [11].
Курсивом обозначается ген, а прямым шрифтом — соответствующий ему белок.
Мутации удобно разделить на несколько классов в соответствии с тем, какие последствия они вызывают (рис. 3) [12].

Рисунок 3. Классы мутаций CFTR при муковисцидозе. GA — аппарат Гольджи; ER — эндоплазматический ретикулум. Красный овал — ядро клетки.
[13], рисунок адаптирован
Мутации I класса
Мутации I класса встречаются примерно у 10% пациентов. При них белок CFTR вообще не синтезируется или синтезируется в усеченном виде и сразу деградирует, потому что в гене произошла замена кодирующего аминокислоту кодона на стоп-кодон, или сдвиг рамки считывания, или появился сигнал неправильного сплайсинга. Самая частая мутация — замена глицина-542 на стоп-кодон.
Мутации II класса
Наиболее распространены мутации II класса, вызывающие неправильные сворачивание белка и последующий процессинг клеточными механизмами. Самая частая мутация — F508del (делеция фенилаланина в положении 508 ). 70% пациентов гомозиготны по этой мутации (то есть она присутствует в обеих копиях гена CFTR), а у 90% есть хотя бы один мутантный аллель [14]. У гомозиготных пациентов наблюдается тяжелое течение муковисцидоза, а гетерозиготы по CFTR-F508del, у которых одна из копий гена нормальна, не имеют симптомов болезни. Существует гипотеза, объясняющая стабильность такого тяжелого заболевания в человеческой популяции: у гетерозигот в меньшей степени происходит потеря воды при болезнях, сопровождающихся диареей, например, при холере и брюшном тифе. Соответственно, раньше, когда эти болезни были одной из основных причин смертности, особенно детской, шел отбор на дефектные копии гена [15].
F — обозначение фенилаланина, а del обозначает делецию, то есть отсутствие аминокислоты.
Мутация F508del приводит к тому, что белок неправильно сворачивается и еще в эндоплазматическом ретикулуме не проходит «контроль качества» со стороны клеточных систем и направляется на деградацию, не доходя до плазматической мембраны.
Впрочем, 1% неправильно свернутого CFTR-F508del все же может достигнуть клеточной поверхности, но там он работает с очень низкой эффективностью из-за того, что мутация нарушает правильную подвижность доменов, необходимую для открывания и закрывания канала [19] (см. врезку). Кроме того, в течение 2,5 минут белок удаляется с поверхности в эндосомы, и там снова решается его судьба: он либо возвращается обратно в плазматическую мембрану клетки, либо уничтожается. Понятно, что большинство мутантных молекул будет при этом уничтожено.
Мутации III класса
Мутации III класса встречаются у 4–5% пациентов и приводят к неправильной регуляции открытия ионного канала. Из них наиболее обычная — G551D, то есть замена глицина в 551 положении домена NBD1 на аспарагиновую кислоту. Она приводит к тому, что канал остается преимущественно закрытым. Появление в этом положении остатка аспарагиновой кислоты с отрицательно заряженной боковой цепью —CH2–COO− препятствует связыванию АТФ и сближению доменов NBD1 и NBD2 из-за отталкивания отрицательных зарядов между аспартатом и фосфатными группами АТФ, а также кислотными остатками домена NBD2 [19].
Мутации IV и других классов
Довольно редкие мутации класса IV (в сумме 1,7% пациентов) приводят к недостаточно сильному току ионов хлора через открытый канал CFTR [12]. Как правило, это замены положительно заряженных остатков аргинина в канале на незаряженные остатки. По-видимому, наличие положительных зарядов в канале необходимо для прохождения через него ионов Cl−. Для больных с этими мутациями характерно довольно легкое течение болезни, зачастую без легочных и панкреатических проявлений.
Некоторые исследователи различают также мутации классов V–VI, при которых производится работающий белок, но в недостаточных количествах, или происходит быстрое удаление CFTR с поверхности клеток. У таких пациентов течение болезни также сравнительно легкое [11].
Почему для муковисцидоза — самого распространенного наследственного заболевания — первые препараты, направленные на молекулярную причину болезни, появились только недавно, в 2012 году? Причин несколько: во-первых, сломать проще, чем починить, поэтому среди лекарств гораздо больше ингибиторов, блокаторов, антагонистов, чем активаторов и агонистов. В случае CFTR требуется восстановить неработающую функцию, что гораздо сложнее. Во-вторых, при других наследственных заболеваниях, обусловленных дефектом одного гена (например, гемофилии или болезни Гоше), пациентам, как правило, помогает введение дефектного белка в виде инъекций. В случае муковисцидоза проблема так просто не решается. CFTR — мембранный белок, и если его просто ввести пациенту, он не попадет в мембрану эпителиальных клеток и не будет выполнять там нужные функции. Делаются попытки разработать генную терапию муковисцидоза, например, доставить ген CFTR в клетки с помощью вирусных частиц, но они пока не увенчались успехом.
А вот малые молекулы, которые были разработаны с учетом сведений о структуре и функции CFTR, уже произвели революцию в лечении муковисцидоза, и, надеемся, это только начало. О них мы и поговорим дальше.
«Калидеко»: первая ласточка
Сразу после открытия факта, что муковисцидоз вызван мутациями в гене CFTR, у исследователей возникло желание синтезировать вещества, которые могли бы хоть отчасти скомпенсировать эффект данных мутаций. Довольно быстро стало понятно, что эти вещества будут грубо делиться по своей функции на два типа — корректоры, которые будут повышать количество дефектного белка (например, с мутацией F508del) на поверхности клетки, и потенциаторы, которые будут усиливать активность белка, уже находящегося на поверхности. Наиболее простой мишенью при этом выглядят мутации класса III: при них белок правильно свернут, находится на мембране, но преимущественно в закрытом состоянии. Поэтому с 2000-х годов начали поиск таких соединений, которые бы повышали вероятность его пребывания в открытом состоянии [22].
Первые несколько классов соединений были либо недостаточно активны, либо малоселективны (то есть связывались и с другими белками), либо обладали неподходящими фармакологическими свойствами (плохая растворимость, стабильность, проникновение в ткани). Наконец в 2009 году, после многочисленных раундов поиска оптимальной структуры, была опубликована структура вещества VX-770 (рис. 5), позже получившего название ивакафтор (торговое наименование — «Калидеко», Kalydeco) [23]. Расскажем немного подробнее обо всём пути разработки этого соединения.

Рисунок 5. Химическая структура ивакафтора («Калидеко», VX-770) (а) и генистеина (б)
История разработки началась в 2000 году. В ту пору компания Aurora Biosciences, которая занималась разработкой систем скрининга для Большой фармы, решила инициировать свою собственную разработку и получила грант от Фонда муковисцидоза (Cystic Fibrosis Foundation). В 2001 году Aurora была куплена компанией Vertex Pharmaceuticals, и та продолжила исследования.
Скрининг — в данном случае процесс выбора нужной молекулы из большого множества.
Для начала потребовалось создать систему скрининга, которая бы подходила для отбора веществ, активных в отношении мутантов CFTR. Для первичного отбора использовали клеточную культуру мышиных фибробластов, синтезирующих мутантный CFTR-F508del. Для определения влияния исследуемой молекулы на хлорный канал ее добавляли к клеточной культуре, затем добавляли флуоресцентный маркер, чувствительный к изменениям мембранного потенциала из-за потока ионов хлора и «включали» CFTR, добавляя форсколин, активирующий канал.
Так было протестировано 228 000 соединений [24], многие из которых отдаленно напоминали генистеин — известный потенциатор CFTR, который, однако, не подходит для лекарственного применения из-за низкой активности и быстрой деградации в организме.
В итоге в 2005 году был синтезирован и отобран VX-770 — он оказался в 2000 раз мощнее генистеина и показал самый долгий период полувыведения у крыс среди всех аналогов (9,5 ч.). Дальнейшее изучение VX-770 показало, что он не связывается с основными цитохромами P450 , не проявляет активности в отношении 160 важных рецепторов, в том числе нервной системы, не ингибирует сердечный калиевый канал hERG. Все это косвенно свидетельствует о приемлемой безопасности лекарственного кандидата, по крайней мере, на данной стадии.
Время, за которое концентрация вещества, введенного в организм, падает в два раза.
Цитохромы P450 — белки печени, которые отвечают за метаболизм многих лекарств, причем у разных людей по-разному. Чем меньше лекарство на них влияет, тем меньше вероятность взаимодействия с другими лекарствами, потенциальная токсичность и вариабельность действия.
Затем VX-770 тщательно изучали in vitro (на изолированных клетках) и in vivo (на животных) [22]. Определили фармакокинетические параметры молекулы у мышей, крыс, собак и обезьян, показали пероральную биодоступность у крыс и собак на уровне 40–50% (то есть что почти половина проглоченного препарата попадает в кровь).
Исследования эффективности на животных моделях для ивакафтора не проводили, хотя, как правило, это необходимо для получения разрешения на клинические исследования. Но в данном случае подходящей модели не было, потому что ивакафтор не связывается с мышиным CFTR, а трансгенных мышей с человеческим белком не было на момент выхода препарата в клинические исследования.
Разработка препарата шла невероятно быстро: уже в 2008 году были опубликованы первые клинические данные для ивакафтора, а к моменту регистрации в 2012 году были доступны данные трех клинических исследований. На 2018 год в пяти двойных слепых клинических исследованиях [25], [26] уже приняли участие 342 пациента. Основным показателем, который изучается у больных муковисцидозом, является FEV1 — объем форсированного выдоха (forced expiratory volume) за одну секунду. Причем измеряется не абсолютный объем, а процент от нормы для данного пола, возраста, расы и роста. Другие важные показатели — безопасность препарата, частота легочных обострений (выражается в необходимости приема антибиотиков и госпитализации) и качество жизни (определяется по анкете, которую заполняет пациент).

Рисунок 6. Ожидаемая продолжительность жизни без муковисцидоза, с муковисцидозом при применении ивакафтора и с муковисцидозом без ивакафтора
[29], рисунок адаптирован
Ивакафтор у детей и взрослых с мутацией G551D через 24 недели приема по сравнению с плацебо улучшал FEV1 на 5–10% и снижал риск обострений на 55%. У взрослых (но не у детей) он также повышал качество жизни по сравнению с плацебо. Воздействие препарата на ионный канал подтверждалось снижением концентрации хлорида в потовой жидкости [27], [28]. Затем ивакафтор показал эффективность и при наличии других мутаций класса III [11]. Немаловажно, что препарат оказался очень безопасным — частота побочных эффектов не превосходила плацебо. К сожалению, в России он так до сих пор и не зарегистрирован.
Хотя пока недостаточно данных для того, чтобы говорить о снижении смертности от муковисцидоза под воздействием ивакафтора, но проведенное моделирование показывает, что его влияние на FEV1 способно вылиться в увеличение продолжительности жизни в среднем на 15 лет (рис. 6), а также снизить потребность в трансплантации легких [29].
Такая эффективность дала основание компании Vertex установить на свой препарат очень высокую цену: годовой курс стоит в США $310 000. Недавние исследования, проведенные в США и Великобритании, показали, что такая цена неоправданно высока [30], [31].
Несмотря на то, что ивакафтор оказался настоящим прорывом в терапии муковисцидоза, его недостаток в том, что он помогает всего 4–5% больных — у пациентов с гомозиготной мутацией F508del он оказался неэффективен [34]. Такой факт мог бы показаться странным, если учесть, что ивакафтор был отобран по активности на клетках с CFTR-F508del. Однако не нужно забывать: эти клетки предварительно выдерживались при комнатной температуре, что помогает правильно свернуться даже мутантному белку, поэтому на их поверхности было достаточно CFTR-F508del.
Комбинации
Понятно, что для охвата большинства пациентов надо создавать молекулы, эффективные у пациентов с мутацией F508del (напомним, они называются корректорами). Компания Vertex взялась за разработку таких препаратов одновременно с потенциаторами. После скрининга 164 000 молекул и нескольких раундов оптимизации было синтезировано вещество VX-809, которое в клеточных тестах в семь раз улучшало созревание CFTR-F508del и в пять — его способность переносить ионы хлора [35]. Позже оно получило название люмакафтор (lumacaftor).
Люмакафтор пробовали применять в режиме монотерапии (без сочетания с другими препаратами), но он оказался малоэффективен. Поэтому Vertex провела клинические исследования комбинации люмакафтор/ивакафтор, которая получила название «Оркамби» (Orkambi), и зарегистрировала ее в 2015 году.
Оказалось, что добавление люмакафтора к ивакафтору несколько улучшало течение болезни у пациентов с гомозиготной мутацией F508del, хотя эффект был не таким сильным, как в случае применения ивакафтора у пациентов с CFTR-G551D. FEV1 улучшался примерно на 5% по сравнению с плацебо, примерно в полтора раза снижалось количество обострений, улучшалось качество жизни [31]. Комбинация оказалась не столь безопасной, как ивакафтор в одиночку: небольшая часть пациентов прекратила прием препарата из-за удушья. Тем не менее эта комбинация позволила охватить около 45% пациентов с муковисцидозом.
Однако параллельно обнаружили, что у пациентов с мутацией F508del ивакафтор вообще несколько снижает эффективность корректоров [36], и, следовательно, требуются новые препараты и комбинации, чтобы повысить эффективность терапии и охватить тех пациентов, кому не подходят существующие лекарства. Кроме того, оказалось, что люмакафтор активирует цитохром CYP3A — белок печени, который отвечает за метаболизм ивакафтора, что снижает эффективность последнего [37].
Компания Vertex не собирается останавливаться: в 2018 году она вывела на рынок новую комбинацию — тезакафтор + ивакафтор («Симдеко», Symdeco). Тезакафтор тоже был обнаружен в ходе высокопроизводительного скрининга 150 000 соединений в 2005 году [38]. В сочетании с ивакафтором на клетках эпителия бронхов, взятых у пациентов с гомозиготной мутацией CFTR-F508del, он продемонстрировал повышение транспорта хлорида до 15,7% от нормального. Также было показано, что комбинация увеличивает частоту биения ресничек эпителия [39].
В клинических исследованиях Symdeco оказался более эффективным, чем один ивакафтор, а главное — комбинация оказалась совсем безопасной: в группе препарата наблюдалось даже меньше нежелательных явлений, чем в плацебо-группе [31].
По структуре тезакафтор похож на люмакафтор, и в обоих случаях точно не известно, где именно они связываются с CFTR и каков механизм компенсации мутации F508del. Бесценные данные по трехмерной структуре CFTR были получены относительно молодым методом криоэлектронной микроскопии, за который в 2017 году присудили Нобелевскую премию по химии [40].
Три зарегистрированных препарата компании Vertex охватывают потребности примерно 60–70% пациентов и уже продаются больше, чем на $2 млрд (данные 2017 года). Однако и это еще не все — с помощью препаратов следующих поколений Vertex намеревается охватить более 90% пациентов. В том же 2017 году компания потратила на исследования $1,32 млрд.
Перспективы
Vertex ведет исследования новых, тройных комбинаций, где к комплексу тезакафтора и ивакафтора будет добавляться еще один корректор — VX-659, или VX-445, или VX-152. Эти три молекулы были идентифицированы в ходе скрининга в присутствии тезакафтора. На клетках комбинация каждого из этих веществ увеличивала поток ионов хлора до 68–75% от нормы [42].
Результаты исследования фазы 3 для первой такой комбинации станут известны в конце 2018 года, для второй — в середине 2019. В фазе 2 увеличение FEV1 по сравнению с плацебо для тройной комбинации тезакафтор + ивакафтор + VX-659 составило 13%, что свидетельствует о довольно высокой эффективности комбинации [41].
На более ранней стадии есть у Vertex и совсем новые разработки — в фазе 2 исследуется комбинация потенциатора, корректора и ингибитора ENaC. Разработчики надеются, что, снижая отток ионов натрия с поверхности эпителиальных клеток внутрь, они добьются еще лучшего восстановления слизистого слоя.
Еще одна интересная разработка — дейтерированный ивакафтор, то есть такой, у которого некоторые атомы водорода заменены на более тяжелый изотоп дейтерий (рис. 9). Это не влияет на активность ивакафтора по отношению к CFTR, но делает его более устойчивым к превращениям в организме. В итоге его период полувыведения примерно в полтора раза длиннее, чем у ивакафтора, что позволяет дозировать его один раз в день вместо двух. Сейчас для дейтерированного ивакафтора идут исследования фазы 2 [43].

Рисунок 9. Дейтерирование молекулы меняет связь углерода с водородом и характер взаимодействия молекулы с некоторыми веществами за счет ослабления водородной связи.
На совсем ранней стадии у Vertex есть и генная терапия, использующая систему CRISPR-Cas9 [45], и мРНК, компенсирующая дефекты CFTR. Однако множество провалов в этой области пока не дают основания утверждать, что именно эти подходы сработают.
Заключение
К сожалению, пока в России не зарегистрировано ни одно из новых средств, описанных в данной статье, но, надеемся, ситуация в ближайшие годы изменится.
На примере муковисцидоза интересно проследить взаимовлияние между разными уровнями организации материи: изменение всего нескольких атомов в молекуле белка CFTR отражается на работе клетки, затем ткани, органа и всего организма. А последствием этого является организация социальных структур из сотен людей, которые предпринимают усилия для компенсации дефекта на уровне молекул.
Вылечить муковисцидоз пока невозможно — для этого требуются более совершенные средства генной терапии, чем у нас есть сейчас. Но описанные в этой статье препараты позволяют значительно улучшить жизнь большинства больных. Главная задача — разработка таких средств, которые бы помогали всем пациентам независимо от мутаций CFTR.
Литература
- CFTR structure and regulation. CFTR.info;
- Кистозный фиброз (муковисцидоз): микробиологическая диагностика хронической респираторной инфекции. (2018). Минздрав РФ;
- Steven V. Molinski, Vijay M. Shahani, Adithya S. Subramanian, Stephen S. MacKinnon, Geoffrey Woollard, et. al.. (2018). Comprehensive mapping of cystic fibrosis mutations to CFTR protein identifies mutation clusters and molecular docking predicts corrector binding site. Proteins. 86, 833-843;
- David C. Gadsby, Paola Vergani, László Csanády. (2006). The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 440, 477-483;
- Bradley S Quon, Steven M Rowe. (2016). New and emerging targeted therapies for cystic fibrosis. BMJ. i859;
- Mark T. Clunes, Richard C. Boucher. (2007). Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder. Drug Discovery Today: Disease Mechanisms. 4, 63-72;
- H. Matsui, M. W. Verghese, M. Kesimer, U. E. Schwab, S. H. Randell, et. al.. (2005). Reduced Three-Dimensional Motility in Dehydrated Airway Mucus Prevents Neutrophil Capture and Killing Bacteria on Airway Epithelial Surfaces. The Journal of Immunology. 175, 1090-1099;
- Richard C. Boucher. (2007). Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends in Molecular Medicine. 13, 231-240;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- Виртуальные тропы реальных лекарств;
- Isabelle Fajac, Claire E. Wainwright. (2017). New treatments targeting the basic defects in cystic fibrosis. La Presse Médicale. 46, e165-e175;
- Michael J. Welsh, Alan E. Smith. (1993). Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 73, 1251-1254;
- Luigi Maiuri, Valeria Raia, Guido Kroemer. (2017). Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 24, 1825-1844;
- Nadia Ameen, Mark Silvis, Neil A. Bradbury. (2007). Endocytic trafficking of CFTR in health and disease. Journal of Cystic Fibrosis. 6, 1-14;
- Xin Meng, Jack Clews, Vasileios Kargas, Xiaomeng Wang, Robert C. Ford. (2017). The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell. Mol. Life Sci.. 74, 23-38;
- Xin Meng, Jack Clews, Eleanor R. Martin, Anca D. Ciuta, Robert C. Ford. (2018). The structural basis of cystic fibrosis. Biochm. Soc. Trans.. 46, 1093-1098;
- Tzyh-Chang Hwang, Jiunn-Tyng Yeh, Jingyao Zhang, Ying-Chun Yu, Han-I Yeh, Samantha Destefano. (2018). Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol.. jgp.201711946;
- Diane E. Grove, Meredith F.N. Rosser, Richard L. Watkins, Douglas M. Cyr. (2011). Analysis of CFTR Folding and Degradation in Transiently Transfected Cells. Methods in Molecular Biology. 219-232;
- Xin Meng, Jack Clews, Eleanor R. Martin, Anca D. Ciuta, Robert C. Ford. (2018). The structural basis of cystic fibrosis. Biochm. Soc. Trans.. 46, 1093-1098;
- GWAS и психогенетика: консорциумы в поисках ассоциаций;
- Sang Hyun Lim, Elizabeth-Ann Legere, Jamie Snider, Igor Stagljar. (2018). Recent Progress in CFTR Interactome Mapping and Its Importance for Cystic Fibrosis. Front. Pharmacol.. 8;
- Sabine Hadida, Fredrick Van Goor, Jinglan Zhou, Vijayalaksmi Arumugam, Jason McCartney, et. al.. (2014). Discovery of N-(2,4-Di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator. J. Med. Chem.. 57, 9776-9795;
- F. Van Goor, S. Hadida, P. D. J. Grootenhuis, B. Burton, D. Cao, et. al.. (2009). Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences. 106, 18825-18830;
- Fredrick Van Goor, Kimberly S. Straley, Dong Cao, Jesús González, Sabine Hadida, et. al.. (2006). Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. American Journal of Physiology-Lung Cellular and Molecular Physiology. 290, L1117-L1130;
- С миру по нитке: как соединились компоненты клинического исследования;
- Путь к тысячам аптек начинается с одной молекулы;
- Peter J Barry, Anna L Donaldson, Andrew M Jones. (2018). Ivacaftor for cystic fibrosis. BMJ. k1783;
- Sanjay Patel, Ian P Sinha, Kerry Dwan, Carlos Echevarria, Michael Schechter, Kevin W Southern. (2015). Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews;
- Piyameth Dilokthornsakul, Ryan N. Hansen, Jonathan D. Campbell. (2016). Forecasting US ivacaftor outcomes and cost in cystic fibrosis patients with the G551D mutation. Eur Respir J. 47, 1697-1705;
- Penny Whiting, Maiwenn Al, Laura Burgers, Marie Westwood, Steve Ryder, et. al.. (2014). Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technology Assessment. 18;
- Modulator treatments for cystic fibrosis: effectiveness and value. (2018). ICER;
- Laura J. Byrnes, Yingrong Xu, Xiayang Qiu, Justin D. Hall, Graham M. West. (2018). Sites associated with Kalydeco binding on human Cystic Fibrosis Transmembrane Conductance Regulator revealed by Hydrogen/Deuterium Exchange. Sci Rep. 8;
- J. P. Clancy. (2014). CFTR Potentiators: Not an Open and Shut Case. Science Translational Medicine. 6, 246fs27-246fs27;
- Patrick A. Flume, Theodore G. Liou, Drucy S. Borowitz, Haihong Li, Karl Yen, et. al.. (2012). Ivacaftor in Subjects With Cystic Fibrosis Who Are Homozygous for the F508del-CFTR Mutation. Chest. 142, 718-724;
- F. Van Goor, S. Hadida, P. D. J. Grootenhuis, B. Burton, J. H. Stack, et. al.. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences. 108, 18843-18848;
- D. M. Cholon, N. L. Quinney, M. L. Fulcher, C. R. Esther, J. Das, et. al.. (2014). Potentiator ivacaftor abrogates pharmacological correction of F508 CFTR in cystic fibrosis. Science Translational Medicine. 6, 246ra96-246ra96;
- Susanna A. McColley. (2016). A safety evaluation of ivacaftor for the treatment of cystic fibrosis. Expert Opinion on Drug Safety. 1-7;
- N. Pedemonte. (2005). Small-molecule correctors of defective F508-CFTR cellular processing identified by high-throughput screening. Journal of Clinical Investigation. 115, 2564-2571;
- Marc A. Sala, Manu Jain. (2018). Tezacaftor for the treatment of cystic fibrosis. Expert Review of Respiratory Medicine. 12, 725-732;
- Крупные подробности микроскопического мира: Нобелевская премия по химии 2017;
- Marjolein Mijnders, Bertrand Kleizen, Ineke Braakman. (2017). Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Current Opinion in Pharmacology. 34, 83-90;
- . (2016). Poster Session Abstracts. Pediatr Pulmonol.. 51, S194-S485;
- Scott L. Harbeson, Adam J. Morgan, Julie F. Liu, Ara M. Aslanian, Sophia Nguyen, et. al.. (2017). Altering Metabolic Profiles of Drugs by Precision Deuteration 2: Discovery of a Deuterated Analog of Ivacaftor with Differentiated Pharmacokinetics for Clinical Development. J Pharmacol Exp Ther. 362, 359-367;
- Стрельцова Ю. (2017). Vertex обзавелась улучшенным «Калидеко» против муковисцидоза. «Мосмедпрепараты».



Комментарии
0Чтобы оставить комментарий, необходимо
войти