Происхождение рака: генетика и эпигенетика
23 января 2026
Происхождение рака: генетика и эпигенетика
- 2059
- 0
- 10
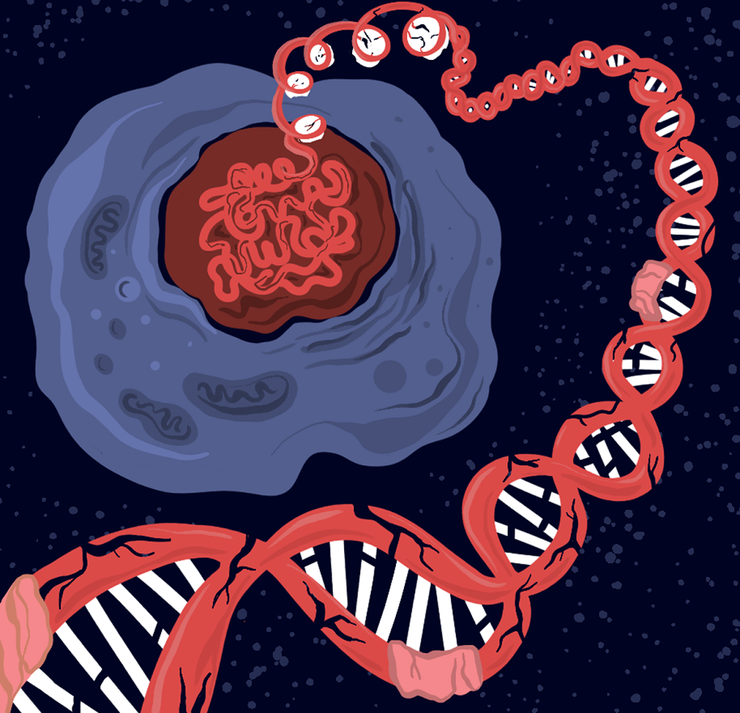
Генетические и эпигенетические изменения — главная движущая сила злокачественного перерождения клеток.
Рисунок в оригинальном разрешении.
иллюстрация Анастасии Самоукиной
-
Автор
-
Редакторы
-
Иллюстратор
Злокачественное перерождение клеток имеет множество аспектов — клеточный, метаболический, иммунологический... Однако начинается все с изменений, происходящих в геноме. Понимание генетических и эпигенетических нарушений в раковых клетках позволяет совершенствовать методы диагностики и таргетной терапии онкологических заболеваний. Продолжаем изучать молекулярные механизмы рака в рамках спецпроекта «Онкология».
Онкология

Онкология — одно из самых динамично развивающихся направлений биомедицинских исследований. С одной стороны медицинское и практическое значение этой темы обеспечивает ей интерес общества и обширное финансирование; с другой — изучение раковой клетки и эволюции опухолей проливает свет на самые фундаментальные вопросы биологии, что делает данный спецпроект интересным для биологов самых разных специальностей.
Редактор спецпроекта — Мария Кондратова, к.б.н специалист в области молекулярной биологии рака и онкоиммунологии, автор научно-популярных книг «Кривое зеркало жизни» и «Невидимый страж», которые мы горячо рекомендуем всем интересующимся молекулярной онкологией и иммунологией.
Партнер спецпроекта — компания SkyGen: передовой дистрибьютор продукции для life science на российском рынке.
Генетическое, но не наследственное!
Чтобы понять природу рака, необходимо начать с причины злокачественного перерождения — изменений, происходящих в геноме раковых клеток. Однако следует понимать, что с функциональной точки зрения геном — очень сложная и запутанная система. В данном очерке мы сможем дать лишь самый общий, и неизбежно фрагментарный обзор того, что происходит с наследственным аппаратом при злокачественном перерождении. Так сказать, попытаемся представить многофигурную «Вавилонскую башню» Брейгеля-старшего в формате почтовой открытки. Однако мы надеемся, что этот текст сможет стать хорошей основой для чтения и понимания более солидных научных источников. Для первого поверхностного вхождения в тему достаточно прочитать основной текст, но тем, кто хочет узнать больше подробностей об особенностях разных онкогенов и онкосупрессоров, мы рекомендуем заглянуть в дополнительные материалы, которые убраны в сворачиваемые врезки. Ну а тех, кто открыл страницы спецпроекта «Онкология» впервые, мы просим сначала прочесть два других материала: «От медицинской онкологии к молекулярной биологии рака» [1] и «Онкодиагностика — вызовы и решения» [2].
В учебниках о злокачественных опухолях часто пишут о раке как о «генетическом заболевании соматических клеток». Такое описание может смутить неспециалиста, привыкшего к тому, что «генетическое заболевание» = «наследственное заболевание». Однако это устаревшее понимание значение термина «генетический». Современная биомедицинская наука использует этот термин во всех случаях, когда причиной заболевания являются те или иные нарушения в геноме человека, независимо от того, наследуются они или нет.
Развитие злокачественных опухолей, как правило, вызывают нарушения в геноме отдельной клетки на протяжении жизни организма, поэтому рак — не наследуется. Небольшая доля опухолей развивается не из соматических клеток, а из половых, — это так называемые тератомы (с греческого «чудовищная опухоль»).
Нельзя унаследовать опухоль, а вот риск ее развития — вполне. Наследственный опухолевый синдром [3] является причиной примерно 5–10% злокачественных опухолей [4] — примерно каждый 50-й человек может быть носителем мутаций, предрасполагающих к развитию опухоли [5]. И поэтому, если в семье были повторяющиеся случаи онкологических заболеваний, особенно одного типа (рак молочной железы) и в раннем (по раковым меркам) возрасте до 60 лет, следует обязательно обратиться к медицинскому генетику. Генетическая предрасположенность к раку — не приговор, но она требует более внимательного отношения к своему здоровью.
Причины возникновения мутаций и эволюция опухолей
Самый простой вариант мутации — ошибка ДНК-полимеразы: ключевого фермента, отвечающего за копирование ДНК. Многие клетки организма регулярно обновляются, и неудивительно, что в копии генома закрадываются ошибки. Они не ограничиваются нуклеотидной заменой, возможен пропуск одной или нескольких «букв». Если кратность пропуска равна трем, то всё может быть еще не так страшно, а в остальных случаях возникает сдвиг рамки считывания, и продукт такого гена совершенно точно не будет правильным.
Случаются и глобальные катастрофы. При репликации каждая ДНК-полимераза копирует лишь часть хромосомы. Если вдруг на каком-то участке необходимый комплекс просто не собирается — целый фрагмент хромосомы просто не реплицируется. В результате там останется одноцепочечная ДНК, которая очень быстро расщепляется нуклеазами, а образовавшийся двухцепочечный разрыв ликвидируется либо гомологичной рекомбинацией [6], либо негомологичным сращиванием концов [7]. В итоге геном теряет значительный фрагмент информации, в который может входить, например, ген или фрагмент гена онкосупрессора.
Аналогично возможна активация онкогена: он может целиком встать под регуляторный элемент другого, активно экспрессирующегося гена; или потерять часть собственной структуры (например, аутоингибиторный домен) — и тогда ему не нужны будут активирующие сигналы. В месте разрыва также может сформироваться химерный ген, «сшитый» из двух кодирующих последовательностей, которые в норме должны экспрессироваться по отдельности. Если разрыв хромосомы произошел в интронах, у такого химерного гена может быть целый ряд различных вариантов активации.
Например, слияние генов (фьюжн) ATG7–RAF1 активирует PI3K/mTOR и MAPK сигнальные пути [8], поскольку RAF1 в такой конструкции постоянно активирован.
Сигнальные пути mTOR и MAPK реагируют на внеклеточные ресурсы и стимулы к делению. В «сытые» времена они блокируют аутофагию и повышают глобальный биосинтез белков. При онкологическом заболевании регуляция этих двух [9], [10] (сразу или по отдельности [11]) сигнальных путей очень часто нарушена.
Химерный ген может возникнуть и в результате реципрокной хромосомной транслокации, при которой происходит обмен фрагментами после двухцепочечных разрывов ДНК. Например, при обмене между 22 и 9 хромосомами формируется фьюжн BCR—ABL1, встречающийся в 95% случаев хронического миелоидного лейкоза . Ген BCR вообще довольно хрупкий, и описано очень много химер с его участием [12]; функциональная активность и драйверный потенциал некоторых из них еще исследуются. Свое название — Breakpoint Cluster Region — он получил из-за того, что разрыв хромосомы образуется именно в этом специфическом и достаточно небольшом участке.
Подробнее об этом заболевании и этом фьюжене, а также о способах борьбы с ними мы уже писали: «От хромосом к молекулам: молекулярная цитогенетика» [13] и «Три поколения лекарств» [14].
Катастрофические события в геноме редко происходят на заре злокачественной трансформации первичной клетки. Для образования опухоли первичной клетке важно отключить онкосупрессоры, и уже после этого в раковых клетках накапливаются мутации, в том числе и «глобальные», затрагивающие целостность хромосомного набора. Хромосомная нестабильность — характерная черта раковых клеточных линий [15]. Но и в организме, чем дальше зашла прогрессия опухоли, тем меньше помех претерпеть хромотрипсию — разрывы и фрагментацию хромосом.
И как всё это жизнеспособно? А никто не говорил, что все раковые клетки выживают. Выживают сильнейшие, и это определяет гетерогенность опухоли, и, следовательно, трудности лечения, но это уже отдельная тема.
Другие события геномного масштаба: инсерция — перемещение участка хромосомы либо внутри хромосомы, либо в другую (внутрихромосомная и межхромосомная транслокации, соответственно); тандемная дупликация — геномный участок дуплицируется и может встать в соседний участок или поменяться с ним местами, как в случае формирования слитого гена FGFR3—TACC3 [16], — в норме ген рецептора фактора роста фибробластов FGFR3 следует за TACC3, но во фьюжене фрагмент из 16 экзонов встает перед 8-м экзоном TACC3, и киназная активность FGFR3 выходит из-под контроля.
Сегменты хромосом могут переворачиваться, и, если гены разнонаправлены, это тоже приводит к формированию химер: например, KIF5B–RET возникает в результате инверсии участка длиной более чем 11 млн пар оснований. Побочно возникает неэкспрессирующийся фьюжн RET–KIF5B.
Наконец, при хромотрипсии хромосомы могут не только распадаться на отдельные фрагменты, но эти фрагменты могут собираться в неправильные конструкции, и тут можно ждать чего угодно [17].
Опухоль, накапливая мутации, развивается эволюционно: в ней происходит отбор жизнеспособных клеток, в чем-то похоже на то, как развивалась жизнь на Земле (рис. 1). Дело тут не только в генетических изменениях: в зависимости от пространственной организации опухоли, на нее действуют разные силы — одни клетки учатся жить в условиях гипоксии; другие, наоборот, находятся близко к сосудам и постоянно взаимодействуют с клетками иммунной системы, учатся «секретным рукопожатиям» [18] (экспрессируют иммунные чекпоинты, такие как PDL-1 или CTLA-4), рекрутируют фибробласты [19], провоцируют дифференцировку Т-хелперов в иммуносупрессивные Т-регуляторные клетки или поляризацию макрофагов из воспалительных М1 в М2, которые не хотят охотиться на злоумышленников, а, наоборот, разрешают заживление ран (читай, деление клеток — рост и развитие опухоли). Третьи клетки выходят в кровоток или лимфоток и формируют колонии сначала в лимфоузлах, а потом и в отдаленных органах; и учатся жить в новых условиях [20].

Рисунок 1. Эволюция и клональная экспансия раковых клеток. Считается, что сóлидные опухоли развиваются годами и десятилетиями. За это время клоны первичной клетки успевают накопить мутации, сформировать колонии, приспособиться к окружающей среде, а отчасти и приспособить под себя окружающую среду. Именно из-за того, что на ранних стадиях опухоль трудно обнаружить, а на поздних она уже сильна, адаптирована и гетерогенна, так важно развивать новые подходы к детекции новообразований за счет специфических маркеров и предупреждать риски с помощью генетических тестов.
иллюстрация Анастасии Самоукиной по [20]
Эволюции способствует нестабильность генома — раковые клетки делятся быстро, часто отключают систему репарации ДНК. Значит, в зависимости от того, на какую часть опухоли мы смотрим, мы будем наблюдать разные изменения в геноме. Раковые клетки будут рождаться и умирать; среда и генетика будут отбирать жизнеспособные клоны. Опухоли не просто разные у разных людей — они неоднородны (гетерогенны) сами по себе. И за те 10–20 лет, что проходят с момента трансформации первичной клетки обнаружения типичной солидной опухоли, произойдет очень много.
Клетка содержит в себе геном из 3,17 млрд «букв». Кодирующая и регуляторная части — в меньшинстве. Еще и мутации должны произойти в нескольких из 500 (а не любом) генов, и не всякие мутации подойдут. Казалось бы, ситуация почти невероятная. Но многие клетки обновляются постоянно — мы как та самая река, в которую нельзя войти дважды: каждый день чуточку новые и другие (слущиваются клетки эпителия, рождаются и вскоре гибнут нейтрофилы, растут волосы и ногти — всего не перечислишь). Каждый день мы подвергаемся воздействию факторов риска: едим пережаренное мясо или пьем алкоголь, курим или вдыхаем накуренное другими, загораем, контактируем с Helicobacter pylori и т.д. Много клеток, много риска, много времени жизни организма: так шанс редкого явления возрастает до знаменитого «дожить до собственного рака».
Генетика опухолей
К счастью, далеко не каждая мутация приводит к формированию злокачественной опухоли. Геном человека избыточен — значительная его часть не кодирует белки и не является регуляторной, и в рамках общепринятой модели онкогенеза мутации в этих — весьма обширных — фрагментах не несут опасности. И среди примерно 20 тысяч генов далеко не каждый, в случае мутации, грозит развитием онкологии.
Мутации, приводящие к злокачественному перерождению клетки, называют «драйверными». Это мутации в генах, продукты которых вовлечены в процесс деления клетки. Одной драйверной мутации недостаточно для зарождения раковой опухоли, но она увеличивает шанс на возникновение второй, третьей... Считается, что три мутации — это минимум для становления онкологического заболевания, однако чаще их требуется больше. И все они должны произойти в одной клетке. Когда клетка преображается в раковую и начинает бесконтрольно делиться, ее геном теряет стабильность, и накопление мутаций ускоряется.
Далеко не каждое изменение в геноме будет способствовать дальнейшему развитию опухоли. Какие-то мутации будут давать опухолевым клеткам преимущество, другие — наоборот, будут их ослаблять или вовсе приводить к гибели, третьи будут нейтральными. Мутации, не способствующие прогрессу и ускорению роста опухоли, называются пассажирскими. Они как бы подсаживаются в эволюционный автобус опухоли, но не влияют на его маршрут, в отличие от драйверных мутаций.
Драйверные мутации происходят в двух типах генов. Первые — протоонкогены — активируют клеточный рост и деление. Например, получают сигнал от факторов роста во внеклеточной среде, или передают этот сигнал дальше по цепочке. Такие гены становятся онкогенами, когда получают усиливающие, активирующие их мутации gain of function — начинают работать неконтролируемо: как если бы ваш телефон звонил без какого бы то ни была сигнала вызова от других абонентов. Беда в том, что каждый такой «звонок» — это сигнал клетке, что пора делиться.
Вторая группа — гены-онкосупрессоры. Их задача в здоровых клетках — проверять, действительно ли есть сигнал к росту и делению (landscapers), контролировать целостность генома (caretakers) и напрямую останавливать деление клетки, если что-то пошло не так (gatekeepers).
«Двухударная модель», предложенная Альфредом Кнудсоном [21], описывает, как онкогены получают контроль над клеткой — оба аллеля гена-онкосупрессора должны потерять функциональную активность, чтобы дать старт развитию опухоли. Кнудсон пришел к этой гипотезе, изучая ретинобластому (опухоль сетчатки) и один из самых известных генов-онкосупрессоров (gatekeeper) — хрестоматийный RB1.
Драйверной мутацией может быть не только изменение кодона, ведущее к аминокислотной замене в белке, но и мутация в регуляторной части: например, оверэкспрессия фактора транскрипции MYC является нарушением, которое с большой вероятностью может привести к неоплазии [22]. Или мутация в промоторе гена TERT (одного из белков теломеразы, достраивающей теломеры на концах хромосом) может провоцировать экспрессию гена и дать клеткам возможность бесконтрольно делиться [23], [24] — преодолеть предел Хейфлика, избежать сенесценции и гибели. Сами белки при этом остаются «нормальными» — в них нет аминокислотных замен или других перестроек, однако в раковых клетках они производятся в заведомо избыточном количестве.
- Предел Хейфлика —
- количество делений, которые может претерпеть здоровая клетка, обычно около 40–60. По мере повторов репликации концы хромосом (теломеры) сокращаются — этим и обусловлен запас делений. Раковые клетки активируют теломеразу, которая также активна в стволовых клетках, и поэтому способны к бесконечной пролиферации.
Активация теломеразы и выход за предел Хейфлика — одна из характерных черт злокачественных клеток (разбирали все такие черты в статье «От медицинской онкологии к молекулярной биологии рака» [1] — обязательно почитайте!). Эта черта, как и многие другие, приобретается в ходе естественного отбора опухолевых клеток, а эволюция — это мутации. Остановимся подробнее на каждой группе генов, вовлеченных в онкогенез.
Протоонкогены и гены-супрессоры опухолей
Итак, пара сотен генов в геноме называются протоонкогенами. «Приобретая функцию», они становятся онкогенами, что неизбежно для многоклеточного организма, в котором обязательно есть системы регуляции роста тканей. Их действие в здоровых клетках уравновешивается активностью генов-супрессоров опухолей. Сигналы внутри клетки чаще всего можно свести к модели баланса между активирующими и ингибирующими — там нет одного «гонца», которого может перехватить один «страж»; скорее, речь будет идти о волне активирующих сигналов, которая сталкивается с волной ингибирующих сигналов, и тут уж кто кого переупрямит. Так, при обнаружении мутации в последовательности ДНК (может быть в протоонкогене) накопление сигнала, ведущего к аресту клеточного цикла, остановит переход в новую фазу. Но не всегда в мутации дело. Существует класс мощных протоонкогенов, способных изменять паттерны экспрессии генов в очень широком диапазоне, и они не требуют мутаций в кодирующей последовательности.

Рисунок 2. Клеточная топография онкогенов и онкосупрессоров. Клетку можно сравнить с мегаполисом — миллионы молекул образуют сложные сети взаимодействий, и когда какая-то группа на генетическом уровне начинает работать слишком активно или, наоборот, теряет бдительность, может произойти «революция», последствия которой могут быть самыми разными — от гибели клетки до бесконтрольного деления и формирования целой колонии таких «революционных» клеток, злонамеренность в которых выходит за пределы молекулярного масштаба и выходит на уровень всего организма.
иллюстрация Анастасии Самоукиной
Факторы транскрипции (ФТ) —
очень могущественная группа генов, чьи продукты прямо или косвенно оказывают влияние на экспрессию сотен и даже тысяч генов в клетке. В сущности, всего четыре ФТ — Oct4, Sox2, Klf4 и c-Myc — способны откатить клетку назад в ее дифференцировке [25] и сделать из какого-нибудь фибробласта индуцированную стволовую клетку, способную переквалифицироваться, например, в нейрон.
Именно за это открытие Синъя Яманака (эти четыре ФТ так и названы — факторы Яманака) и Джон Гердон получили в 2012 году Нобелевскую премию по физиологии и медицине [26], [27]. И один из этих факторов — c-Myc — входит в топ самых мощных онкогенов [28]. Ему даже не нужна мутация — достаточно нарушенного баланса экспрессии. Вместе со «звездой» молекулярной онкологии p53 это — один из наиболее часто активируемых онкогенов среди человеческих злокачественных опухолей.
Регуляторы клеточного цикла
Циклин-зависимые киназы (CDK) регулируют переход из одной фазы клеточного цикла в другую, и как с такими полномочиями не быть протоонкогенами [32]? Касается это не только «классических» CDKs (CDK1, 2, 4, 6): есть целый ряд других CDK, которые могут играть онкогенную или онкосупрессивную роль. А некоторые (CDK8, 12, 19) — сразу обе.
Рецепторы факторов роста
Просто так клетки организма не растут. Процесс этот регулируется сразу несколькими способами: в самом общем виде клетка должна получить сигнал о том, что надо расти, что есть за счет чего расти, и не должно быть сигнала о том, что расти некуда. «Надо расти» — это сигналы от факторов роста (их в человеческом организме много: EGF — эпидермальный фактор роста, FGF — фактор роста фибробластов, PDGF — тромбоцитарный фактор роста, и так далее). В большинстве случаев сигнал факторов роста принимают рецепторные тирозинкиназы [37]. У этих трансмембранных рецепторов всего одна трансмембранная ɑ-спираль: сигнал внутрь клетки передается за счет димеризации рецепторов (рис. 3) [38]. При онкогенных мутациях для димеризации и последующего аутофосфорилирования никакие сигналы уже не нужны.

Рисунок 3. Схема димеризации рецепторных тирозинкиназ в норме. Сложность разработки специфических ингибиторов рецепторных тирозинкиназ состоит в том, что это огромный класс молекул, очень похожих друг на друга. Наиболее разительные отличия — в сайте связывания лиганда, но беда в том, что онкогенные РТК уже не нуждаются в нем, чтобы димеризоваться, фосфорилировать друг друга и таким образом передать сигнал; а вот сайты связывания АТФ, которая служит донором для сигнального фосфата, или сайты фосфорилирования очень похожи, и создать молекулу, которая могла бы заблокировать одну РТК, не затронув работу других, очень непростая задача.
иллюстрация Анастасии Самоукиной по [38]
G-белки
Семиспиральные GPCR (G-белоксопряженные рецепторы) после связывания с лигандом изменяют взаимную конформацию своих семи ɑ-спиралей, и это приводит к активации G-белка (бывают гетеротримерные и «малые»: и те, и другие имеют ГТФазную активность, необходимую для инактивации). Им помогают белки GEF и GAP: соответственно, заменяют «холостую» молекулу ГДФ на «боевую» ГТФ и позволяют отщипнуть от нее фосфат. Но G-белок — не киназа. Замена ГДФ на ГТФ осуществляется для активации G-белка: он меняет конформацию и может связаться и активировать Raf-белок [46]. Если в результате мутации G-белок теряет ГТФазную активность, он остается в активной конформации.
Безостановочные сигналы к пролиферации создают большую нагрузку на репликационную машину. Мутация ДНК-полимеразы приводит к лавинообразному накоплению мутаций [53]. Например, в CpG-островках, которые часто бывают метилированы по цитозину, тот часто заменяется на тимин, очень похожий на 5-метилцитозин. Как бы то ни было, множественные нарушения целостности генома, а также стресс, который испытывает клетка с нарушенными сигнальными путями и метаболизмом, должны бы привести ее к апоптозу.
Но не тут-то было. Апоптоз, видимо, часто случается на ранних стадиях развития опухоли, как фактор отбора: выживают те, кто научился его избегать: активирующие мутации в антиапоптотическом гене Bcl-2 — весьма ценное приобретение для опухолевых клеток [54].
Онкосупрессоры
Онкогенные мутации обладают большим потенциалом, но было бы оскорбительно в отношении эволюции думать, что за долгие годы не нашлось, что противопоставить разрушительному потенциалу мутаций в протоонкогенах (рис. 4). В современной научной литературе выделяют три типа генов-онкосупрессоров. Разделение весьма условно. Gatekeepers — гены-привратники — контролируют клеточный цикл и проверяют, насколько правомерен переход к следующей фазе. Caretakers — смотрители, стражники генома; их задача — остановить процесс деления, если в геном вкралась ошибка, исправить ее, а если это невозможно — запустить апоптоз. Сравнительно недавно была сформулирована третья группа — landscapers. Они контролируют внеклеточный матрикс, следят за сигналами внеклеточной среды.

Рисунок 4. Клеточный цикл и фаза действия некоторых онкогенов и онкосупрессоров. Знание о том, в какой фазе клеточного цикла работает онкоген, может помочь при разработке таргетной терапии, потому что это показывает, каким образом онкоген обходит все защитные системы клетки, призванные уберечь ее от неконтролируемого деления и злокачественного перерождения. Вдобавок, это может иметь и прогностическую ценность: как правило, онкогены, активные в S-, а также G2/M-фазах, сопряжены с более высоким риском геномной нестабильности и худшим прогнозом.
иллюстрация Анастасии Самоукиной
Эпигенетические эффекты в опухолях
В ландшафт раковых клеток сложности (казалось бы, куда больше?) добавляет эпигенетика [66] — система генетического контроля, лежащая «поверх» генетики (с греческого «эпи-» — поверх, над): изменения в работе генов, не связанные с изменением последовательности ДНК. В случае злокачественных опухолей правильнее дополнить: «не связанные с изменением в их последовательности ДНК», потому что мутации всё же присутствуют. «Биомолекула» выпускала Спецпроект по эпигенетике, куда мы и направляем читателей, чтобы познакомиться с этой концепцией подробнее: «Молекулы и эпигеном» [67], а также «Эпигеном: параллельная реальность внутри клетки» [68].
Эпигенетические изменения могут влиять на развитие клеток, их дифференцировку, адаптацию к сигналам окружающей среды, системы общения с соседними и отдаленными клеткам (паракринные и эндокринные сигналы).
В случае злокачественной трансформации эпигенетика вторична по отношению к генетическим нарушениям. Однако, постойте. Теоретически возможен эпигенетически спровоцированный рак: метилирование ДНК или транскрипция некодирующих РНК подавляет экспрессию онкосупрессоров — накопление мутаций уже вторично. Было показано, что гиперметилирование промотора p16 приводило к более раннему развитию спонтанных опухолей у мышей [69]. А репрессия ремоделлера хроматина из семейства Polycomb привела к развитию исключительно эпигенетически спровоцированного рака у дрозофилы [70]. Однако это искусственно созданные «в тепличных условиях» опухоли.
Есть три базовых варианта взаимовлияния генетики и эпигенетики на рост и развитие опухоли [71]:
- Мутантные факторы транскрипции привлекают кофакторы (активаторы и репрессоры), факторы ремоделирования хроматина и другие белковые комплексы, перестраивающие укладку хроматина и меняющие паттерны экспрессии генов.
- Мутации в генах факторов ремоделинга хроматина ведут к тому, что они изменяют свою активность или работают менее специфично.
- Мутации в регуляторных последовательностях генов не дают факторам транскрипции с ними связаться и так нарушают программы их экспрессии.
Недавно группа из Южной Кореи обнаружила три регуляторных фактора [72]: гистон деацетилазу (HDAC2) и два ФТ: MYB и FOXA2, ингибирование которых позволило дифференцировать опухолевые клетки колоректального рака в нормальные энтероциты. Это открытие открывает новые перспективы разработки эпигенетической терапии рака.
Заключение: зачем искать новые драйверные мутации?
Надеюсь, после прочтения этой статьи у вас не осталось сомнений, что именно мутации определяют поведение опухоли. С развитием технологий секвенирования [104], то есть прочтения последовательности молекул ДНК и РНК, получение информации о мутациях опухоли и уровне экспрессии генов стало рутинной задачей. И если полногеномное секвенирование пока еще дорого, то полнотранскриптомное (то есть, совокупность РНК) дешевле, и позволяет довольно точно оценить, активен ли мутантный ген.
Проводятся также диагностические операции солидных опухолей, и полученный образец, после проведения исследования тканей, позволит определить степень дифференцировки клеток, состав микроокружения опухоли, ее инвазивность.
Хорошо известная мутация в гене BRAF V600E увеличивает активность белка в среднем в 480 раз [105]. Эта мутация чаще всего встречается в колоректальном раке [106], меланоме, раке щитовидной железы и немелкоклеточном раке легкого. А замена N581S всего-навсего в шесть раз активнее. Замена валина в 600 положении на глутамат, аспартат, лизин и аргинин и встречается гораздо чаще, поэтому сейчас в клинике есть целый арсенал из ингибиторов BRAF V600E/D/K/R: вемурафениб, дабрафениб, энкорафениб.
Вообще, если в названии лекарства в конце есть «-иб» — значит, это ингибитор. И вот целый ряд таких молекул (олопариб, нарипариб, талазопариб и рукапариб) применяются в случаях, когда наследственный опухолевый синдром всё же привел к развитию рака. У этих ингибиторов есть еще кое-что общее в названии — все они -парибы, то есть ингибиторы PARP. Это поли(АДФ-рибоза)-полимеразы, которые, что для нас наиболее важно здесь, вовлечены в процессы репарации ДНК. Ингибиторы чаще всего нацелены на PARP1 [107], который считается наиболее важным в сигнализации об одно- и двухцепочечных разрывах ДНК. Таргетная терапия против PARP часто сопровождает химиотерапию, и логика здесь следующая: если у человека наследственная мутация в одном из генов репарации ДНК (CHEK1/2, PALB2, ATM, CDK12, RAD51C, BRIP1, PPP2R2A, RAD51D, FANCL, RAD51B, RAD54L — кстати, на целостность и правильность этих генов можно провериться), то, вызывая повреждения ДНК в раковых клетках с помощью «химии» и блокируя того, кто будет об этих повреждениях сигналить, можно убивать опухолевые клетки более эффективно.
Для клинического использования одобрены и многие другие молекулы, блокирующие активность мутантных белков. И не только мутантных — есть средства, нарушающие полимеризацию тубулина, то есть ломающие цитоскелет, каркас клетки, или ингибирующие протеасому — такие клетки не могут поддерживать гомеостаз, потому что разобрать белки и собрать из деталей новые чуть ли не важнее, чем получить детали извне.
В заключение надо добавить, что далеко не все ученые согласны, что рак — это генетическое заболевание [108]. Возникла идея назвать его метаболическим заболеванием [109]. Чего больше в этом желании сменить знамена: революционного духа несоглашательства, своего ракурса взгляда на рак или желания создать сенсацию? Ракурс действительно важен: объять всю литературу невозможно, рассредоточить собственные исследования по невообразимо сложному ландшафту — тем более. А своя область, естественно, дороже и важнее.
Метаболизм действительно важен [110] — если бы проблема опухоли включала в себя только злокачественные клетки, жизнь онкологов — как врачей, так и ученых — была бы намного проще, а жизнь пациентов — скорее всего, дольше. Но злосчастное микроокружение опухоли [111] — клетки, которые его формируют, их поведение, дифференцировка или поляризация — во многом зависят от метаболизма. Среда, в которой оказываются клетки, влияет на эпигенетическую регуляцию транскриптома, и поведение микроокружения опухоли — один из важнейших прогностических и диагностических факторов в новых терапевтических практиках.
Однако какой бы теоретический и практический интерес не представляли эти новаторские взгляды на природу рака, на сегодняшний день именно изучение геномных и эпигеномных изменений в опухоли продолжает оставаться основным источником диагностических и терапевтических разработок, позволяющих спасти жизни множеству больных. Генетика рака продолжает оставаться динамичной развивающейся областью науки и обещает нам еще много удивительных открытий.
Литература
- От медицинской онкологии к молекулярной биологии рака;
- Онкодиагностика — вызовы и решения;
- Mattia Garutti, Lorenzo Foffano, Roberta Mazzeo, Anna Michelotti, Lucia Da Ros, et. al.. (2023). Hereditary Cancer Syndromes: A Comprehensive Review with a Visual Tool. Genes. 14, 1025;
- Van Thuan Tran, Sao Trung Nguyen, Xuan Dung Pham, Thanh Hai Phan, Van Chu Nguyen, et. al.. (2022). Pathogenic Variant Profile of Hereditary Cancer Syndromes in a Vietnamese Cohort. Front. Oncol.. 11;
- Evgeny N Imyanitov, Ekaterina S Kuligina, Anna P Sokolenko, Evgeny N Suspitsin, Grigoriy A Yanus, et. al.. (2023). Hereditary cancer syndromes. World J Clin Oncol. 14, 40-68;
- L. Krejci, V. Altmannova, M. Spirek, X. Zhao. (2012). Homologous recombination and its regulation. Nucleic Acids Research. 40, 5795-5818;
- Eric Weterings, David J Chen. (2008). The endless tale of non-homologous end-joining. Cell Res. 18, 114-124;
- Poonam Sonawane, Patrick Diviney, Joanna Phillips, Payal Jain, Phillip Storm, et. al.. (2018). LGG-57. FUNCTIONAL CHARACTERIZATION AND EFFECTIVE TARGETING OF NRF1-BRAF AND ATG7-RAF1 FUSIONS IDENTIFIED IN ANAPLASTIC PLEOMORPHIC XANTHOASTROCYTOMA PATIENTS WITHOUT BRAF p.V600E MUTATION. Neuro-Oncology. 20, i116-i116;
- David A. Guertin, David M. Sabatini. (2007). Defining the Role of mTOR in Cancer. Cancer Cell. 12, 9-22;
- A S Dhillon, S Hagan, O Rath, W Kolch. (2007). MAP kinase signalling pathways in cancer. Oncogene. 26, 3279-3290;
- Diana-Theodora Morgos, Constantin Stefani, Daniela Miricescu, Maria Greabu, Silviu Stanciu, et. al.. (2024). Targeting PI3K/AKT/mTOR and MAPK Signaling Pathways in Gastric Cancer. IJMS. 25, 1848;
- Malalage N. Peiris, Fangda Li, Daniel J. Donoghue. (2019). BCR: a promiscuous fusion partner in hematopoietic disorders. Oncotarget. 10, 2738-2754;
- От хромосом к молекулам: молекулярная цитогенетика;
- Три поколения лекарств;
- Haomin Huang, Michael Lampson, Andrey Efimov, Timothy J. Yen. (2018). Chromosome instability in tumor cells due to defects in Aurora B mediated error correction at kinetochores. Cell Cycle. 17, 2622-2636;
- Devendra Singh, Joseph Minhow Chan, Pietro Zoppoli, Francesco Niola, Ryan Sullivan, et. al.. (2012). Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science. 337, 1231-1235;
- Paul A. Northcott, David J. H. Shih, John Peacock, Livia Garzia, A. Sorana Morrissy, et. al.. (2012). Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 488, 49-56;
- Клеточная мафия и коррумпированные комиссары;
- Lea Monteran, Neta Erez. (2019). The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol.. 10;
- Arghavan Ashouri, Chufan Zhang, Federico Gaiti. (2023). Decoding Cancer Evolution: Integrating Genetic and Non-Genetic Insights. Genes. 14, 1856;
- Alfred G. Knudson. (1971). Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. U.S.A.. 68, 820-823;
- Renumathy Dhanasekaran, Anja Deutzmann, Wadie D. Mahauad-Fernandez, Aida S. Hansen, Arvin M. Gouw, Dean W. Felsher. (2022). The MYC oncogene — the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol. 19, 23-36;
- Franklin W. Huang, Eran Hodis, Mary Jue Xu, Gregory V. Kryukov, Lynda Chin, Levi A. Garraway. (2013). Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science. 339, 957-959;
- Susanne Horn, Adina Figl, P. Sivaramakrishna Rachakonda, Christine Fischer, Antje Sucker, et. al.. (2013). TERT Promoter Mutations in Familial and Sporadic Melanoma. Science. 339, 959-961;
- Kazutoshi Takahashi, Shinya Yamanaka. (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 126, 663-676;
- Была клетка простая, стала стволовая;
- Нобелевская премия по физиологии и медицине (2012): индуцированные стволовые клетки;
- Zachary E. Stine, Zandra E. Walton, Brian J. Altman, Annie L. Hsieh, Chi V. Dang. (2015). MYC, Metabolism, and Cancer. Cancer Discovery. 5, 1024-1039;
- Douglas Hanahan. (2022). Hallmarks of Cancer: New Dimensions. Cancer Discov. 12, 31-46;
- Болезни и изменения клеточного метаболизма;
- Jim Battey, Christopher Moulding, Rebecca Taub, William Murphy, Timothy Stewart, et. al.. (1983). The human c-myc oncogene: Structural consequences of translocation into the igh locus in Burkitt lymphoma. Cell. 34, 779-787;
- Циклины и их помощники — регуляторы клеточного цикла;
- Ilenia Pellarin, Alessandra Dall’Acqua, Andrea Favero, Ilenia Segatto, Valentina Rossi, et. al.. (2025). Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Sig Transduct Target Ther. 10;
- Dazhi Xu, Chien-Feng Li, Xian Zhang, Zhaohui Gong, Chia-Hsin Chan, et. al.. (2015). Skp2–MacroH2A1–CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat Commun. 6;
- Ioana I. Nitulescu, Sara C. Meyer, Qiang Jeremy Wen, John D. Crispino, Madeleine E. Lemieux, et. al.. (2017). Mediator Kinase Phosphorylation of STAT1 S727 Promotes Growth of Neoplasms With JAK-STAT Activation. EBioMedicine. 26, 112-125;
- Henry E. Pelish, Brian B. Liau, Ioana I. Nitulescu, Anupong Tangpeerachaikul, Zachary C. Poss, et. al.. (2015). Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 526, 273-276;
- Deric L. Wheeler, Yosef Yarden Receptor Tyrosine Kinases: Family and Subfamilies — Springer International Publishing, 2015;
- Ciprian Tomuleasa, Adrian-Bogdan Tigu, Raluca Munteanu, Cristian-Silviu Moldovan, David Kegyes, et. al.. (2024). Therapeutic advances of targeting receptor tyrosine kinases in cancer. Sig Transduct Target Ther. 9;
- Gur Pines, Wolfgang J. Köstler, Yosef Yarden. (2010). Oncogenic mutant forms of EGFR: Lessons in signal transduction and targets for cancer therapy. FEBS Letters. 584, 2699-2706;
- Gilda da Cunha Santos, Frances A. Shepherd, Ming Sound Tsao. (2011). EGFR Mutations and Lung Cancer. Annu. Rev. Pathol. Mech. Dis.. 6, 49-69;
- Justyna Rawluk, Cornelius F. Waller. (2018). Gefitinib. Recent Results in Cancer Research. 235-246;
- Gregory J. Riely, Helena A. Yu. (2015). EGFR: The Paradigm of an Oncogene-Driven Lung Cancer. Clinical Cancer Research. 21, 2221-2226;
- Rolando Perez, Tania Crombet, Joel de Leon, Ernesto Moreno. (2013). A view on EGFR-targeted therapies from the oncogene-addiction perspective. Front. Pharmacol.. 4;
- I. Bernard Weinstein, Andrew Joe. (2008). Oncogene Addiction. Cancer Research. 68, 3077-3080;
- Toril Holien, Anders Sundan. (2012). Oncogene addiction to c-MYC in myeloma cells. Oncotarget. 3, 739-740;
- Christian Johnson, Deborah L. Burkhart, Kevin M. Haigis. (2022). Classification of KRAS -Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discovery. 12, 913-923;
- Ji Luo. (2021). KRAS mutation in pancreatic cancer. Seminars in Oncology. 48, 10-18;
- Niki Karachaliou, Clara Mayo, Carlota Costa, Ignacio Magrí, Ana Gimenez-Capitan, et. al.. (2013). KRAS Mutations in Lung Cancer. Clinical Lung Cancer. 14, 205-214;
- Ингибиторы Ras: в поисках Грааля таргетной терапии;
- Что мы узнали о биологии рака из клинических исследований. Несколько новелл об онкологии;
- Kathryn E. Mercer, Catrin A. Pritchard. (2003). Raf proteins and cancer: B-Raf is identified as a mutational target. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1653, 25-40;
- Cornelia Braicu, Mihail Buse, Constantin Busuioc, Rares Drula, Diana Gulei, et. al.. (2019). A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers. 11, 1618;
- Marketa Tomkova, Michael John McClellan, Gilles Crevel, Akbar Muhammed Shahid, Nandini Mozumdar, et. al.. (2024). Human DNA polymerase ε is a source of C>T mutations at CpG dinucleotides. Nat Genet. 56, 2506-2516;
- J M Adams, S Cory. (2007). The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 26, 1324-1337;
- William Hankey, Wendy L. Frankel, Joanna Groden. (2018). Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: implications for therapeutic targeting. Cancer Metastasis Rev. 37, 159-172;
- Jiaqi Liu, Qing Xiao, Jiani Xiao, Chenxi Niu, Yuanyuan Li, et. al.. (2022). Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Sig Transduct Target Ther. 7;
- Важнейшие стрелочники клеток организма: белки Wnt;
- Ming Zhao, Lopa Mishra, Chu-Xia Deng. (2018). The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci.. 14, 111-123;
- Päivi Peltomäki. (2005). Lynch Syndrome Genes. Familial Cancer. 4, 227-232;
- C A Cremona, A Behrens. (2014). ATM signalling and cancer. Oncogene. 33, 3351-3360;
- Mark J. O’Connor. (2015). Targeting the DNA Damage Response in Cancer. Molecular Cell. 60, 547-560;
- Cyriac Kandoth, Michael D. McLellan, Fabio Vandin, Kai Ye, Beifang Niu, et. al.. (2013). Mutational landscape and significance across 12 major cancer types. Nature. 502, 333-339;
- Rui Kamada, Yu Toguchi, Takao Nomura, Toshiaki Imagawa, Kazuyasu Sakaguchi. (2016). Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers. 106, 598-612;
- Konstantinos Voskarides, Nefeli Giannopoulou. (2023). The Role of TP53 in Adaptation and Evolution. Cells. 12, 512;
- S.-Y. Shieh. (1999). DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. The EMBO Journal. 18, 1815-1823;
- Cathérine Dupont, D. Armant, Carol Brenner. (2009). Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin Reprod Med. 27, 351-357;
- Молекулы и эпигеном ;
- Эпигеном: параллельная реальность внутри клетки;
- Da-Hai Yu, Robert A. Waterland, Pumin Zhang, Deborah Schady, Miao-Hsueh Chen, et. al.. (2014). Targeted p16Ink4a epimutation causes tumorigenesis and reduces survival in mice. J. Clin. Invest.. 124, 3708-3712;
- V. Parreno, V. Loubiere, B. Schuettengruber, L. Fritsch, C. C. Rawal, et. al.. (2024). Transient loss of Polycomb components induces an epigenetic cancer fate. Nature. 629, 688-696;
- Félix Recillas-Targa. (2022). Cancer Epigenetics: An Overview. Archives of Medical Research. 53, 732-740;
- Jeong‐Ryeol Gong, Chun‐Kyung Lee, Hoon‐Min Kim, Juhee Kim, Jaeog Jeon, et. al.. (2025). Control of Cellular Differentiation Trajectories for Cancer Reversion. Advanced Science. 12;
- Dou-Sheng Bai, Chi Zhang, Ping Chen, Sheng-Jie Jin, Guo-Qing Jiang. (2017). The prognostic correlation of AFP level at diagnosis with pathological grade, progression, and survival of patients with hepatocellular carcinoma. Sci Rep. 7;
- Hui Shen, Peter W. Laird. (2013). Interplay between the Cancer Genome and Epigenome. Cell. 153, 38-55;
- Winston Timp, Andrew P. Feinberg. (2013). Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 13, 497-510;
- Douglas E. Laux, Edward M. Curran, Wade V. Welshons, Dennis B. Lubahn, Tim H.‐M. Huang. (1999). Hypermethylation of the Wilms' tumor suppressor gene CpG island in human breast carcinomas. Breast Cancer Res Treat. 56, 35-43;
- Matteo Forloni, Romi Gupta, Arvindhan Nagarajan, Li-Sha Sun, Yuying Dong, et. al.. (2016). Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Reports. 16, 457-471;
- Srinivasan Yegnasubramanian, Jeanne Kowalski, Mark L. Gonzalgo, Marianna Zahurak, Steven Piantadosi, et. al.. (2004). Hypermethylation of CpG Islands in Primary and Metastatic Human Prostate Cancer. Cancer Research. 64, 1975-1986;
- Verena Sailer, Ulrike Sailer, Emma Grace Bawden, Romina Zarbl, Constanze Wiek, et. al.. (2019). DNA methylation of indoleamine 2,3-dioxygenase 1 (IDO1) in head and neck squamous cell carcinomas correlates with IDO1 expression, HPV status, patients’ survival, immune cell infiltrates, mutational load, and interferon γ signature. EBioMedicine. 48, 341-352;
- Christiane A. Opitz, Luis F. Somarribas Patterson, Soumya R. Mohapatra, Dyah L. Dewi, Ahmed Sadik, et. al.. (2020). The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer. 122, 30-44;
- Adam Kinnaird, Steven Zhao, Kathryn E. Wellen, Evangelos D. Michelakis. (2016). Metabolic control of epigenetics in cancer. Nat Rev Cancer. 16, 694-707;
- Hetakshi Kurani, Seyedeh Fatemeh Razavipour, Kuzhuvelil B. Harikumar, Matthew Dunworth, Andrew J. Ewald, et. al.. (2022). DOT1L Is a Novel Cancer Stem Cell Target for Triple-Negative Breast Cancer. Clinical Cancer Research. 28, 1948-1965;
- Alamelu Nachiyappan, Neelima Gupta, Reshma Taneja. (2022). EHMT1/EHMT2 in EMT, cancer stemness and drug resistance: emerging evidence and mechanisms. The FEBS Journal. 289, 1329-1351;
- Myoung Sook Kim, Ho Jeong Kwon, You Mie Lee, Jin Hyen Baek, Jae-Eun Jang, et. al.. (2001). Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 7, 437-443;
- Paulina Miziak, Marzena Baran, Lidia Borkiewicz, Tomasz Trombik, Andrzej Stepulak. (2024). Acetylation of Histone H3 in Cancer Progression and Prognosis. IJMS. 25, 10982;
- Pingping Guo, Wenqi Chen, Huiyu Li, Meiying Li, Lisha Li. (2018). The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications. Pathol. Oncol. Res.. 24, 807-813;
- Albert M. Li, Jiangbin Ye. (2024). Deciphering the Warburg Effect: Metabolic Reprogramming, Epigenetic Remodeling, and Cell Dedifferentiation. Annual Review of Cancer Biology. 8, 35-58;
- Вослед Варбургу — последние достижения в изучении биоэнергетики рака;
- Метаболизм и эпигеном;
- Meng-Xi Liu, Lei Jin, Si-Jia Sun, Peng Liu, Xu Feng, et. al.. (2018). Metabolic reprogramming by PCK1 promotes TCA cataplerosis, oxidative stress and apoptosis in liver cancer cells and suppresses hepatocellular carcinoma. Oncogene. 37, 1637-1653;
- Ji Hye Kim, Jinyoung Lee, Se Seul Im, Boyun Kim, Eun-Young Kim, et. al.. (2024). Glutamine-mediated epigenetic regulation of cFLIP underlies resistance to TRAIL in pancreatic cancer. Exp Mol Med. 56, 1013-1026;
- Обо всех РНК на свете, больших и малых;
- Olga Y. Burenina, Roman K. Makarchenko, Maria P. Rubtsova, Olga A. Dontsova. (2025). Liver‐specific
lncRNAs associated with liver cancers. FEBS Open Bio. 15, 1383-1405; - Wanxu Huang, Hua Li, Qingsong Yu, Wei Xiao, Dan Ohtan Wang. (2022). LncRNA-mediated DNA methylation: an emerging mechanism in cancer and beyond. J Exp Clin Cancer Res. 41;
- Felipe C. Beckedorff, Murilo Sena Amaral, Carlos Deocesano-Pereira, Sergio Verjovski-Almeida. (2013). Long non-coding RNAs and their implications in cancer epigenetics. Bioscience Reports. 33;
- Кочующие трикстеры и охота на них;
- Zhonghua Ma, Hesuyuan Huang, Yetao Xu, Xuezhi He, Jirong Wang, et. al.. (2017). Current advances of long non-coding RNA highly upregulated in liver cancer in human tumors. OTT. Volume 10, 4711-4717;
- Olga Y. Burenina, Natalia L. Lazarevich, Inna F. Kustova, Daria A. Shavochkina, Ekaterina A. Moroz, et. al.. (2021). Panel of potential lncRNA biomarkers can distinguish various types of liver malignant and benign tumors. J Cancer Res Clin Oncol. 147, 49-59;
- Hyuna Sung, Jacques Ferlay, Rebecca L. Siegel, Mathieu Laversanne, Isabelle Soerjomataram, et. al.. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J Clinicians. 71, 209-249;
- Lianglu Zhang, Lanlan Dong, Changming Lu, Wenxian Huang, Cuiping Yang, et. al.. (2021). Methylation of SDC2/TFPI2 and Its Diagnostic Value in Colorectal Tumorous Lesions. Front. Mol. Biosci.. 8;
- Thomas F. Imperiale, David F. Ransohoff, Steven H. Itzkowitz, Theodore R. Levin, Philip Lavin, et. al.. (2014). Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N Engl J Med. 370, 1287-1297;
- Weisong Zhang, Chaogang Yang, Shuyi Wang, Zhenxian Xiang, Rongzhang Dou, et. al.. (2021). SDC2 and TFPI2 Methylation in Stool Samples as an Integrated Biomarker for Early Detection of Colorectal Cancer. CMAR. Volume 13, 3601-3617;
- Zhijie Wang, Zixuan He, Rong Lin, Zhijie Feng, Xiuling Li, et. al.. (2024). Evaluation of a plasma cell-free DNA methylation test for colorectal cancer diagnosis: a multicenter clinical study. BMC Med. 22;
- 12 методов в картинках: секвенирование нуклеиновых кислот;
- Paul T.C Wan, Mathew J Garnett, S.Mark Roe, Sharlene Lee, Dan Niculescu-Duvaz, et. al.. (2004). Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell. 116, 855-867;
- Jeff Owsley, Matthew K Stein, Jason Porter, Gino K In, Mohamed Salem, et. al.. (2021). Prevalence of class I–III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp Biol Med (Maywood). 246, 31-39;
- Vanessa Patel, Sandra Casimiro, Catarina Abreu, Tiago Barroso, Rita Teixeira de Sousa, et. al.. (2024). DNA damage targeted therapy for advanced breast cancer. Exploration of Targeted Anti-tumor Therapy. 5, 678-698;
- David S. Wishart. (2015). Is Cancer a Genetic Disease or a Metabolic Disease?. EBioMedicine. 2, 478-479;
- Thomas N Seyfried, Laura M Shelton. (2010). Cancer as a metabolic disease. Nutr Metab (Lond). 7, 7;
- Hilary A. Coller. (2014). Is Cancer a Metabolic Disease?. The American Journal of Pathology. 184, 4-17;
- Опухолевые разговоры, или Роль микроокружения в развитии рака.



Комментарии
0Чтобы оставить комментарий, необходимо
войти