Молодость в энергии
01 марта 2021
Молодость в энергии
- 15107
- 0
- 12
Старение митохондрий — один из ключевых компонентов старения организмов. Изучить этот процесс — значит оказаться на шаг ближе к преодолению возрастных заболеваний!
-
Авторы
-
Редакторы
Статья на конкурс «Био/Мол/Текст»: В 1973 году Феодосий Добжанский опубликовал работу, в которой прозвучала ставшая крылатой фраза: «Ничто в изучении биологии не имеет смысла кроме как в свете эволюции». Немного перефразируя Добжанского, можно с большой долей уверенности сказать: «ничто в изучении долголетия не имеет смысла, кроме как в свете митохондриальной медицины». Действительно, состояние митохондрий очень хорошо отражает общее состояние организма. Давайте же поговорим о том, какие болезни могут возникнуть в результате нарушения работы митохондрий, как их можно исправить и зачем это нужно.

Конкурс «Био/Мол/Текст»-2020/2021
Эта работа опубликована в номинации «Свободная тема» конкурса «Био/Мол/Текст»-2020/2021.
Генеральный партнер конкурса — ежегодная биотехнологическая конференция BiotechClub, организованная международной инновационной биотехнологической компанией BIOCAD.

Спонсор конкурса — компания SkyGen: передовой дистрибьютор продукции для life science на российском рынке.
Спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
«Книжный» спонсор конкурса — «Альпина нон-фикшн»
О митохондриях и их функциях
Нет такой возрастной патологии, в которой бы не играла ключевую роль митохондриальная дисфункция. Если ваши митохондрии нормально обновляются и не продуцируют излишне много активных форм кислорода (АФК) — значит, скорее всего, вы молоды, здоровы и счастливы.
Когда-то давно, много тысяч лет назад, человек приручил диких предков собак, сделав их своими помощниками и защитниками. Что-то похожее, только на микроуровне, случилось в процессе эволюции более миллиарда лет назад. Дело в том, что эукариотические клетки возникли в процессе объединения двух более простых клеток: крошечных альфа-протеобактерий, которые стали митохондриями, и архей (локиархей, согласно некоторым данным) [1–3]. Что заставило эти два организма слиться в симбиозе, пока не до конца ясно. Вероятно, они обменивались друг с другом метаболитами, отчего оба получали выгоду. И лишь потом главной функцией митохондрий стал синтез энергии в виде АТФ. Ученые и сейчас расходятся во мнении, было ли изначально это сожительство взаимовыгодным, или же предки митохондрий попали «в рабство». Как бы то ни было, этот симбиоз позволил клетке эукариот подняться на новый энергетический уровень, сделав возможным появление многоклеточных организмов во всем их нынешнем многообразии [4], [5]. Так что же такое митохондрии? Еще в школе мы учили, что клетку можно представить, как целое государство, а митохондрии в ней — «энергетические станции», которые обеспечивают запасание энергии в виде АТФ [6]. Сейчас наше понимание роли митохондрий существенно расширилось.
Сегодня ученые утверждают (и у нас нет повода им не доверять), что митохондрии — это биоэнергетический, биосинтетический и сигнальный центр клетки. Если обобщить, то вот какие функции они выполняют:
- энергетическую;
- сигнальную;
- синтетическую;
- участвуют в процессе запрограммированной клеточной гибели — апоптозе;
- иммунную.
Митохондрии (рис. 1) — полуавтономные органеллы клетки. Они имеют две мембраны и собственный наследственный материал в виде кольцевой молекулы ДНК. ДНК митохондрий содержит всего 37 генов и кодирует 13 белков, что составляет около 1% от их общей потребности. Именно наличие собственного генетического материала — главное свидетельство «бактериального прошлого» митохондрии. Остальные гены в ходе длительного существования внутри клетки «перебрались» в ядро, и ядерная ДНК обеспечивает митохондрии подавляющим количеством белков, необходимых для их работы. Митохондрии нужны белки, которые кодируются и ядерной, и собственной ДНК, поэтому для нормального функционирования клетки необходимо безоговорочное «взаимопонимание» между ядром и митохондриями. Поэтому митохондрии способны как анализировать сигналы, поступающие от ядра, так и передавать ему сигналы в процессе ретроградного сигналинга. Важную роль в коммуникации ядра и митохондрий играют свободные радикалы, но об этом мы поговорим чуть позже, а сначала — об основных функциях митохондрий.

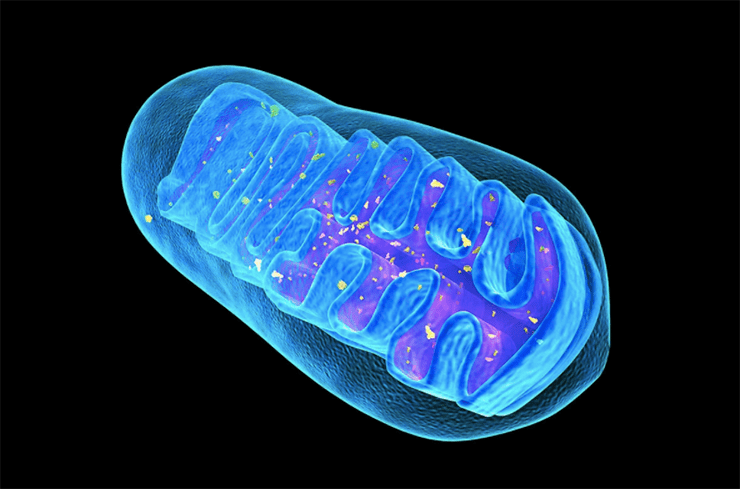
Рисунок 1. Строение митохондрии. Помимо собственной ДНК, в митохондриях есть две мембраны — внешняя и внутренняя. Внутренняя мембрана образует выросты — кристы, — и именно на ней расположена дыхательная цепь, которая обеспечивает запасание энергии в виде молекулы АТФ.
Энергии не занимать!
Из курса физики мы знаем, что ток — это направленный поток электронов. Дыхательная цепь митохондрий представляет собой цепь переноса электронов (рис. 2). Только вместо проводов во внутренней мембране расположена целая система трансмембранных белков (комплексов дыхательной цепи) и специальных переносчиков электронов, которые обеспечивают их направленный поток. Представить этот процесс можно в виде передачи пожарными друг другу ведер с водой. При этом комплексы и переносчики расположены таким хитрым образом, что при переходе электрона от одного к другому происходит выделение энергии в цепи окислительно-восстановительных процессов. Примерно, как если бы ведро с водой пожарные передавали своим товарищам, стоящим ниже на ступеньках (согласитесь, это гораздо легче, чем передавать его вышестоящим). Во внутренней мембране одной митохондрии находятся десятки тысяч полных дыхательных цепей.

Рисунок 2. Дыхательная цепь митохондрий. Всего различают четыре комплекса дыхательной цепи, имеющих довольно сложные названия: НАДН-дегидрогеназа (I), сукцинатдегидрогеназа (II), цитохром-С-оксидоредуктаза (III) и цитохром-С-оксидаза (IV). Электрон передается от комплекса I или II к комплексу III, а от него — к комплексу IV. Передача электрона происходит при помощи специальных переносчиков, самым известным из которых это кофермент Q.
Откуда берется электрон, который «путешествует» по цепи переносчиков внутренней мембраны митохондрий? А берется он из водорода. Если помните, водород — очень простой атом. Он состоит всего из одного протона и одного электрона. Водород мы получаем из молекул, которые потребляем с пищей. Будь то жирные кислоты или глюкоза — в конечном счете, после ряда биохимических процессов и превращений, специальный переносчик (НАДН или ФАДН2) доставляет к дыхательной цепи заветный атом водорода, который даст протон Н+ и электрон.
Куда девается электрон, прошедший по цепи? Конечный его акцептор — кислород. Именно поэтому наше дыхание — кислородное. К слову сказать, есть и другие способы дыхания (например бескислородные). Тогда электрон переносится не на кислород, а на другой акцептор [7].
Казалось бы, всё просто? Так куда же девается протон (Н+), когда электрон отправляется в путешествие по цепи переносчиков? Этот вопрос долго не беспокоил ученых.
Длительное время после открытия комплексов дыхательной цепи тайна дыхания казалась разгаданной. Химия тогда была в большом почете, окислительно-восстановительные реакции активно изучались. Только одно оставалось неясным — переносчик электрона к АТФ-синтазе так и не был обнаружен. И только хемиосмотическая теория, предложенная Питером Митчеллом, пролила свет на тайну дыхания и показала, что такого переносчика просто не существует, а в образовании энергии ключевую роль играет не химия ферментов, а физика мембран.
АТФ-синтаза — еще один компонент внутренней мембраны митохондрий. Это настоящий наномоторчик, который на фотографиях электронного микроскопа и на молекулярных моделях (рис. 3) напоминает гриб. Именно в результате работы АТФ-синтазы образуются молекулы АТФ. Как же это происходит? Та энергия, которая образовалась в окислительно-восстановительных реакциях переноса электронов, идет вовсе не на синтез АТФ, а на «выкачивание» протонов Н+ в пространство между двумя мембранами митохондрий. Когда их там оказывается достаточно, мембрана становится заряженной, как аккумулятор. То есть на внутренней мембране митохондрии возникает электрохимический градиент (он же — мембранный потенциал). Вернуться внутрь митохондрии протоны могут только через АТФ-синтазу (видео 1).

Рисунок 3а. Строение АТФ-синтазы. АТФ-синтаза представляет собой комплекс ферментов, объединенных в несколько субъединиц.

Рисунок 3б. Кристаллографическая модель Ca-АТФ-синтазы
Видео 1. Работа электрон-транспортной цепи и АТФ-синтазы. Представьте себе, как вода, падая с высоты, вращает колесо гидротурбины. Нечто похожее происходит и тут: протоны «вращают» ротор АТФ-синтазы. У людей для каждого полного оборота головки нужны девять протонов; при этом образуются три молекулы АТФ.
Вернемся ненадолго к электронам, которые движутся по дыхательной цепи. Поток электронов нельзя останавливать. Если в работе дыхательной цепи что-то идет не так (плохо «отработал» переносчик электронов, поступление субстрата в виде атомов водорода превышает расход АТФ), то дыхательная цепь может застопориться. Это схоже с работой конвейера: если какое-то звено работает плохо, нарушается весь процесс. Чаще всего «сбоят» первый и третий комплексы. Снижение их активности, к сожалению, происходит и при старении. В результате электроны «выпрыгивают» из дыхательной цепи, а поскольку поблизости всегда есть кислород, электроны «цепляются» к нему, образуя супероксид-анион, О2−. Это так называемый свободный радикал — частица с неспаренным электроном. Существует и еще более вредоносный гидроксильный радикал — ОН−. Он образуется в результате реакции Фентона: Fe2+ + H2O2 → Fe3+ + OH· + OH−.
Неспаренный электрон — это всегда неспокойно. Он постоянно находится в поисках пары, поэтому пытается ее отобрать у других молекул, приводя к образованию всё новых свободных радикалов, которые могут повреждать макромолекулы (ДНК, липиды, белки).
Вам письмо!
Хотя митохондрии называют полуавтономными органеллами, в их ДНК закодировано всего 13 белков. Поэтому они должны обмениваться с ядром сигналами, говорящими об их потребностях и влияющими на активность генов. Ретроградный сигналинг, который мы уже упоминали, активно изучают, ведь его нарушение приводит к поломкам митохондрий и последующему старению или гибели клетки.
На каком же языке «разговаривают» митохондрии? Существует несколько путей коммуникации митохондрий с ядром. Мы приведем лишь некоторые из них:
- Активные формы кислорода (АФК), о причинах образования которых мы уже сказали. Нейтрализацией АФК занимаются системы антиоксидантной защиты, расположенные как в митохондриях, так и в цитоплазме клетки. Раньше считалось, что АФК — исключительно вредный, побочный продукт работы митохондрий, приводящий к старению и гибели клетки [8]. Но это, прежде всего, сигнал, «красная лампочка», которая говорит о проблемах в дыхательной цепи. Возможно, ядру нужно запустить синтез белков для комплексов дыхательной цепи и усилить системы антиоксидантной защиты. Кроме особенностей работы дыхательной цепи на образование АФК существенным образом влияет доступность кислорода. Поэтому эти молекулы первыми запускают каскады адаптации к условиям гипоксии. На этом многообразие сигнальных функций АФК не заканчивается: они принимают участие в регулировании аутофагии [9], [10], иммунных процессов, пролиферации и дифференцировки стволовых клеток и т.д. [11].
- Протеостаз — это баланс синтеза и распада белков [12]. Белки составляют основу нашей жизни, поэтому повреждение белков и утрата гомеостаза белков приводит к ответной стрессовой реакции клетки. В клетке это чаще всего происходит в эндоплазматической сети. Однако нарушение протеостаза в митохондриях также приводит к стрессу.
Именно нарушение протеостаза — основа интереснейшего явления митогормезиса — воздействия на митохондрии по принципу «всё что нас не убивает, продлевает нам жизнь». Использование таких антибиотиков как доксициклин и хлорамфеникол ингибирует синтез белков митохондриями (ведь они похожи на бактерии). При этом снижается уровень митохондриального дыхания и образования АФК и активируются процессы восстановления протеостаза. У C. elegans такой подход продлевает жизнь [13]. Поэтому «мягкое» нарушение работы митохондрий часто только стимулирует их. Немного неудовлетворенные митохондрии посылают сигналы тревоги в ядро, призывая к активации протеостаза. Воздействие на митохондриальный протеостаз сейчас активно изучается в связи с болезнями старения. - Метаболиты цикла Кребса влияют на метилирование и ацетилирование ДНК, то есть являются эпигенетическими модификаторами жизнедеятельности клетки [11], [14], [15].
Помимо коммуникации с ядром у митохондрий есть и другие сигнальные функции. Например, антивирусный сигналинг. При этом митохондрии выступают в роли «датчика вторжения» при попадании вируса в клетку [16]. Но об этом мы поговорим дальше.
Синтезировали-синтезировали...
Помимо синтеза АТФ, в митохондриях происходит еще много всего интересного. Метаболиты цикла Кребса, протекающего в митохондриях, являются предшественниками биосинтеза многих макромолекул, включая липиды, углеводы, белки и нуклеотиды. Митохондрии также участвуют в образовании белков, содержащих гемовые и порфириновые мотивы, и еще в начальных этапах синтеза стероидных гормонов [11], [17].
Эффектное самоубийство
Вероятно на этапах становления симбиоза, если что-то начинало идти не так, митохондрии могли покинуть клетку-хозяина, попросту убив ее. Учитывая тот факт, что у всех многоклеточных организмов митохондрии участвуют в апоптозе, в это нетрудно поверить [18].
Апоптоз — процесс программируемой клеточной гибели [19]. Исходно вызванная предками митохондрий гибель клетки хозяина, вероятно, более напоминала паразитизм и уж никак не приносила одноклеточному хозяину пользу. Но для поддержания нормальной физиологии многоклеточного организма апоптоз просто необходим. Он играет важную роль в развитии и функционировании иммунной системы, гомеостазе и дифференцировке клеток. Его нарушение приводит к онкологическим и нейродегенеративным заболеваниям. У позвоночных апоптоз начинается с изменения проницаемости внешней мембраны митохондрий и высвобождения цитохрома С, активирующего ферменты каспазы. Каспазы же запускают сложную машину программируемой клеточной гибели. К увеличению проницаемости мембраны митохондрий могут приводить как повреждения внутри самой митохондрии, так и вне нее [16].
Подспорье на баррикадах иммунной борьбы
Кроме производства энергии и функций, которые мы уже обсудили, митохондрии исполняют роль «цепного пса» иммунитета — помогают клеткам противостоять атакам бактерий, вирусов и грибов. Кроме этого, митохондрии следят «за порядком в доме». При нахождении клеточных повреждений митохондрии активируют врожденные иммунные реакции, в том числе воспаление. Так организм защищается от внешней и внутренней опасности, реагируя на нарушение гомеостаза.
В зависимости от того, что запускает реакцию врожденного иммунитета — вторжение чужеродного агента или внутренние повреждения, — различают две модели его активации. Во время инфекции врожденный иммунитет активируется, распознавая патоген-ассоциированные молекулярные паттерны (pathogen associated molecular patterns, PAMP) — фрагменты клеточной стенки бактерий, нуклеиновые кислоты бактерий и вирусов и др. Эти паттерны распознаются особыми рецепторами в клетках иммунной системы — PRR (pattern recognition receptors) — своеобразными «датчиками вторжения», которые запускают воспалительный иммунный ответ. К ним относятся толл-подобные рецепторы (TLR), рецепторы RIG-I (retinoic acid-inducible gene-I-like receptors) и MDA-5 (melanoma differentiation-associated protein 5). TLR-рецепторы расположены на мембране клеток или в внутри них, и способны распознавать бактерии, вирусы и грибы [20], [21]. Рецепторы RIG-I и MDA-5 находятся исключительно в цитоплазме и включаются в работу после попадания вируса внутрь клетки. И те, и другие узнают чужеродные агенты по структурным и генетическим компонентам.
При травмах, повреждении тканей и гибели клеток в кровь попадают схожие с PAMP молекулярные фрагменты, которые также запускают иммунный ответ. Они носят название DAMP (damage associated molecular patterns) — например, фибриноген, гистоны или мтДНК.
Атака на вирусы
Однако кроме мембранных и цитоплазматических рецепторов в клетке есть еще активные игроки иммунной защиты — митохондрии. На их внешней мембране локализован белок MAVS (mitochondrial antiviral signaling), работающий «датчиком вторжения» патогена и реагирующий на чужеродный наследственный материал — ДНК и РНК вирусов (рис. 4) [16], [22]. Интересно, что некоторые вирусы (например, гепатитов A, B и C) приспособились «атаковать» MAVS при помощи ферментов — протеаз [23].

Рисунок 4. Механизм действия белка MAVS. Распознав патоген, MAVS меняет конформацию и запускает сигнальный каскад, стимулирующий производство интерферонов типа I и других провоспалительных цитокинов для предотвращения репликации вирусов.
Роль митохондрий в противовирусной защите этим не ограничивается. Многие вирусы, попадая в клетку, воздействуют на митохондрии и их динамику (в норме митохондрии всё время делятся и сливаются) и приводят к дисфункции этих органелл (рис. 5). Есть разные мнения насчет того, делают ли они это в своих «корыстных» целях, или же это происходит по выработанному эволюцией механизму, поскольку нередко для вирусов такая стратегия оказывается губительной.

Рисунок 5. Воздействие вирусов на митохондрии при вторжении в клетку. Вирус SARS-CoV имеет фактор вирулентности ORF-9b, который приводит к деградации DRP1 (белка, который отвечает за фрагментацию митохондрий) и к гиперслиянию митохондрий. Белок ВИЧ gp120 также вызывает снижение уровня DRP1. Вирус лихорадки Денге фактически растворяет митохондриальную мембрану. Вирусные белки вначале колонизируют мембрану эндоплазматического ретикулума, формируя каркас, необходимый для репликации РНК вируса, а затем вступают в контакт с находящейся рядом мембраной митохондрий и делают в ней «дырку». Это ведет к нарушению нормальной работы митохондрий, повышению уровня АФК, дальнейшему повреждению мембраны митохондрий и утечке мтДНК в цитоплазму.
[24], рисунок адаптирован
Почему же такая стратегия часто вредит вирусам? Она вызывает утечку мтДНК в цитоплазму. Это, с одной стороны, приводит к митохондриальной дисфункции, но с другой — стимулирует противовирусный иммунитет (рис. 6).

Рисунок 6. Механизмы, активируемые при разрушении митохондрий. Разрушение митохондрий вызывает выход мтДНК в цитоплазму и запускает иммунный ответ через путь cGAS-STING. Это компонент врожденной иммунной системы, который позволяет обнаружить ДНК в цитоплазме и в ответ запускает экспрессию провоспалительных генов. ДНК распознается особым белком — циклической ГМФ-АМФ синтазой (cGAS), которая запускает превращение ГМФ и АМФ в 2′3′-цГАМФ. Он, в свою очередь, связывается со стимулятором генов интерферонов (stimulator of interferon genes, STING) и активирует каскад фосфорилирования транскрипционных факторов, что приводит к синтезу интерферонов I типа.
Таким образом, митохондрии, как истинные «охранные псы», включают клеточную сигнализацию, жертвуя собой.
И бактерии тоже победим!
Митохондрии реагируют на вторжение в клетку не только вирусов. Они, некогда сами будучи бактериями, могут противостоять вторжению своих «диких сородичей».
Митохондрии участвуют в таком необычном способе защиты от патогенов, как «внеклеточные ловушки». Этот механизм был открыт в 2004 году учеными Института инфекционной биологии имени Макса Планка (Берлин, Германия) (рис. 7а). Кроме того, мтДНК играет важную роль в «системе оповещения» о внедрении патогена. Как уже было сказано выше, она представляет собой один из DAMP и связывается с рецептором TLR9 внутри клетки. Воздействие патогена стимулирует выделение внеклеточных везикул с мтДНК (рис. 7б), в результате чего активируются антибактериальные и провоспалительные сигнальные пути.

Рисунок 7а. При вторжении патогена нейтрофилы могут выбрасывать ДНК (ядерную и митохондриальную), которая формирует во внеклеточном пространстве сетеподобные структуры — нейтрофильные внеклеточные ловушки. Такая «ловушка» служит каркасом для антимикробных медиаторов, таких как белки гранул иммунных клеток, и участвует в процессах врожденного иммунитета [25–27].

Рисунок 7б. мтДНК, заключенная в везикулы, или даже целые митохондрии, попадает в соседние клетки, передавая таким образом сигнал о вторжении патогена в организм
Интересным способом противостоят митохондрии вторжению токсоплазмы — паразитического внутриклеточного простейшего. Токсоплазма при попадании в клетку запускает липофагию — аутофагию липидных капель, в результате которой ликвидируются жирные кислоты. Чтобы перекрыть токсоплазме доступ к жирным кислотам, митохондрии сливаются друг с другом, «захватывая» липиды и окисляя их [28].
Повреждение митохондрий при вторжении патогенов, сопровождающееся выделением мтДНК и увеличением производства АФК, — мощный стимулятор воспаления. Но совсем иначе складывается ситуация, когда врожденный иммунитет стимулируют не вторгшиеся патогены, а клеточный «мусор». Тогда аномальное накопление поврежденных митохондрий и высвобождение мтДНК в цитозоль становятся причиной различных воспалительных заболеваний и усиления хронического возрастного воспаления. Об этом мы и поговорим далее.
Воспаление и митохондрии
Мы уже упоминали, что помимо компонентов вирусов и бактерий воспаление могут запускать молекулярные фрагменты собственных клеток.
Необходимо помнить, что компоненты митохондрий являются достаточно мощными DAMPs. Ученые полагают, что это связано с бактериальным прошлым митохондрий. Пока митохондрии здоровы и находятся внутри клетки, окруженные двойной мембраной, они не привлекают к себе никакого внимания иммунитета. Но как только мембрана митохондрий повреждается, а мтДНК выходит из митохондрии, запускается воспалительная реакция. Какие же компоненты митохондрий относятся к DAMP?
- Кардиолипин — фосфолипид, который у нас и у других эукариот в норме находится только во внутренней мембране митохондрий, где он и синтезируется. Также он часто встречается среди компонентов мембран бактерий (тест на наличие антител к кардиолипину проводят, например, при подозрении на сифилис).
- Формил-пептиды. Синтез белков в митохондриях, как и у бактерий, всегда начинается с особой модифицированной аминокислоты — N-формилметионина. У нас эта аминокислота при синтезе белка не используется. Поэтому наличие N-формилметионина на конце белковой молекулы (или на концах более коротких молекул — формил-пептидов, образующихся при распаде бактериальных белков) — надежный индикатор присутствия бактерий в организме.
- АТФ — универсальный источник энергии для всех биохимических процессов, протекающих в клетке. Мы обычно считаем, что чем АТФ больше — тем лучше. На самом деле свободной АТФ в единичный момент времени в клетке находится очень мало. Бóльшая часть этой молекулы связана и находится в комплексе с магнием.
- TFAM. Ген TFAM находится в ядре. А вот его продукт — белок TFAM — быстро переносится в митохондрии, где плотно связывается с мтДНК, регулируя ее транскрипцию и репликацию. При повреждениях митохондрий и последующем апоптозе клеток он «отцепляется» от мтДНК и активирует иммунные клетки, связываясь с RAGE (receptor for advanced glycation end products) — рецептором конечных продуктов гликирования. RAGE играют важнейшую роль в процессе воспаления [29] и могут активироваться как конечными продуктами гликирования, так и другими сигналами, такими как TFAM.
- Митохондриальная ДНК (мтДНК), вне всякого сомнения, один из важнейших DAMP, поскольку очень похожа на бактериальную ДНК. Она также кольцевая, и тоже содержит характерные «бактериальные последовательности» — неметилированные участки соседствующих нуклеотидов — цитозина и гуанина (CpG-сайты). В нашем ядерном геноме, как и у других млекопитающих, такие участки ДНК обычно подвергаются метилированию, поэтому появление мтДНК в крови воспринимается иммунной системой как опасность.
Роль митохондрий в запуске воспаления не ограничивается тем, что врожденный иммунитет воспринимает их компоненты как DAMPs. Важно помнить, что мтДНК и кардиолипин приводят к активации сборки инфламмасомы (рис. 8).

Рисунок 8. Активация инфламмасомы. NLRP3-инфламмасома обнаруживает микробные вещества и стимулирует «стерильное воспаление», возникающее без воздействия внешних факторов. При отсутствии угрозы в клетке инфламмасома находится в «разобранном состоянии». Молекулярный путь ее активации проходит в два этапа — активация (осуществляется нашими знакомыми сигналами PAMP или DAMP) и сборка. Инфламмасома запускает превращение прокаспазы-1 в каспазу-1 (протеолитический фермент, играющий важнейшую роль в воспалении) и последующую секрецию провоспалительных цитокинов IL-1β и IL-18. После активации IL-1β способствует проникновению иммунных клеток в инфицированные или поврежденные ткани, а IL-18 необходим для выработки интерферона-гамма, который стимулирует клетки иммунной системы. Также каспаза-1 запускает пироптоз. Для этого она расщепляет «белок смерти» газдермин D. Активированный газдермин D перфорирует клеточную мембрану, клетка разбухает и гибнет. Дестабилизация митохондрий, высвобождение мтДНК и кардиолипина являются важными факторами активации инфламмасомы.
К сожалению, повреждение митохондрий происходит не только при вторжении патогенов, когда оно выполняет защитную роль. Митохондрии неизбежно выходят из строя при старении, запуская «стерильное воспаление». Сегодня этот хронический процесс считается общей и одной из главных особенностей старения тканей и причиной большинства возрастных заболеваний [31], [32].
Стареют не только клетки!
Итак, процессы, происходящие в митохондриях, играют важную роль в старении. И если раньше внимание было приковано только к АФК, то сегодня понимание роли митохондрий в старении существенно расширилось (рис. 9) [33].

Рисунок 9. Роль митохондрий в старении клетки и организма
Видео 2. Почему возникают повреждения в митохондриях. Энергетика клеток, биоэнергетическая адаптация клеток и причины их нарушения. Митохондрии — клеточные сенсоры питательных веществ. Здоровая конкуренция субстратов в митохондриях — залог метаболического здоровья. Митохондрии стволовых и сенесцентных клеток, в чем их особенности. Роль митохондрий в стерильном воспалении и аутоиммунитете. Как связано старение организма и работа митохондрий.
С каждым годом к уже описанным процессам прибавляются всё новые и новые, что расширяет наше понимание ситуации. С возрастом происходят следующие изменения, затрагивающие митохондрии [34–36]:
- снижается общее их количество;
- происходит нарушение их внутренней структуры;
- снижается общее количество мтДНК;
- накапливаются различные повреждения мтДНК;
- нарушаются процессы контроля качества митохондрий — митофагии (удаления поврежденных митохондрий), биогенеза (образования митохондрий), динамики митохондрий.
Отчего же возникает митохондриальная дисфункция?
Повреждения мтДНК называли одной из главных причин старения еще со времен господства свободнорадикальной теории старения Хармана. Правда тогда считали, что главная и единственная причина возникновения мутаций — свободные радикалы. В ходе клеточного дыхания митохондриям всё время приходится иметь дело с АФК. Логично предположить, что такая вредная работа со временем приводит к мутациям в мтДНК — резервы истощаются. Сейчас накоплены данные, что существенная часть мутаций мтДНК вызвана ошибками репликации, а не окислительными повреждениями [37]. Как бы то ни было, частота мутаций мтДНК увеличивается с возрастом как у животных, так и у человека. При этом количество копий мтДНК, несущих мутацию, и вид мутаций различаются между тканями и внутри них. Количество мутаций мтДНК растет с возрастом в мозге, сердце, толстом кишечнике, скелетной мускулатуре. Возникают они и в стволовых клетках.
Существует предположение, что повышенное количество мутаций мтДНК влияет на старение и возрастные заболевания. Возражения скептиков сводятся к тому, что вряд ли они достигают такого уровня, чтобы ускорять старение, потому что мтДНК существует в сотнях тысяч копий на клетку. Для того чтобы клетка по-настоящему начала страдать от митохондриальной дисфункции, количество «поломанных» митохондрий должно преодолеть некую «пороговую черту» — около 60% всех митохондрий клетки должны стать нерабочими. Но любая ткань состоит из множества клеток! Сколько же митохондрий должно сломаться, чтобы действительно толкнуть процессы старения?
У сторонников же важной роли поломок мтДНК есть свой аргумент. Генетически модифицированные мыши с дефектной митохондриальной ДНК-полимеразой POLGγ имели сниженное количество копий мтДНК и накапливали ее мутантную форму. И одновременно развивали ускоренный фенотип старения! У них наблюдалось раннее облысение и морщинистость кожи. При этом, если у мышей восстанавливали нормальную работу фермента, то «старческий» фенотип обращался вспять, и мышь снова начинала выглядеть как молодая [38]. Неожиданное объяснение этому явлению было предложено в работе 2019 года А. Суомалайнен. Оказывается, митохондрии, испытывая «дефицит» копий своей ДНК, могут попросту «воровать» нуклеотиды, предназначенные для ядерной ДНК. При этом в мтДНК возникают повреждения, так как нарушаются процессы ее починки [39].
Кроме этого, старение сопровождается нарушением процесса «контроля качества» митохондрий (mitochondrial quality control, MQC). MQC контролирует процессы упаковки и деградации белка, биогенез митохондрий, их динамику и митофагию. Нарушение этих процессов при старении приводит к накоплению поврежденных митохондрий, высвобождению DAMP, окислительному стрессу и стерильному воспалению [40].
Можно ли вылечить митохондрии?
В связи с влиянием процессов, протекающих в митохондриях, на здоровье человека появилось такое направление, как митохондриальная медицина . Это комплекс мероприятий, направленных на улучшение митохондриальной функции не только при митохондриальных заболеваниях (так называемой первичной митохондриальной дисфункции), но и при возрастзависимых заболеваниях. О том, какие патологии связаны с митохондриальными нарушениями, и что об этом думают современные ученые, отлично рассказано на сайте «Биомолекулы» в спецпроекте «Биоэнергетика» [41–43].
Термин «митохондриальная медицина» изначально возник в связи с исследованиями наследственных митохондриальных заболеваний. Сейчас это понятие расширилось и на возрастную дисфункцию митохондрий.
Митохондриальная медицина развивается!
А сейчас мы попробуем структурировать основные подходы митохондриальной медицины:
- Применение антиоксидантов.
- Митогормезис — запуск механизмов адаптации в ответ на стрессовое воздействие. При этом наблюдается адаптивное улучшение работы митохондрий.
- Стимуляция биогенеза митохондрий и процессов их динамики.
- Стимуляция митофагии.
- Оптимизация электрон-транспортной цепи митохондрий.
- Стабилизация липидов их внутренней мембраны.
- Трансплантация митохондрий.
- Генное редактирование митохондриальной ДНК.
Так как процессы, протекающие в митохондриях, взаимосвязаны, данные направления могут пересекаться. Например, стабилизация мембран митохондрий повышает качество процессов митофагии.
Кроме того, направления митохондриальной медицины можно подразделить на:
- диагностику функций митохондрий;
- консервативные подходы (изменение образа жизни);
- инновационные подходы (трансплантацию митохондрий, генную терапию митохондрий, перенос их генов в ядро и др.).
Видео 3. Понятие митохондриальной медицины. Методы диагностики дисфункции митохондрий. Генетические маркеры дисфункции. Методы консервативного поддержания работы митохондрий: спорт, питание, питательные вещества, антиоксиданты и другие подходы.
А ну-ка все на диагностику!
Качественная и достоверная диагностика митохондриальной дисфункции необходима и для разработки терапий, и для оценки их эффективности. Между тем, в этой области не существует стандартного набора методов. Зато существует ряд трудностей:
- Наличие «пороговой черты». В клетке находится множество митохондрий, и только часть из них может быть дисфункциональной. Чтобы в клетке нарушилась митохондриальная функция, доля «больных» митохондрий должна преодолеть некоторую пороговую черту. Считается, что для нарушения работы клетки «нерабочими» должны стать около 60–80% митохондрий. До этого накопление дисфункциональных митохондрий никак внешне не проявляется, а значит, окно возможностей терапии сужается [37], [45].
- Гетероплазмия. Так как клетки содержат разное количество митохондрий в зависимости от уровня метаболической активности ткани, в которой они находятся, генетический материал в этих митохондриях может быть различным. Некоторые из них могут нести мутацию, а другие — нет. Это явление называется гетероплазмией и существенно затрудняет генетическую диагностику наследственного материала митохондрий.
- Тканеспецифичность. Различные ткани имеют разную степень выраженности митохондриальной дисфункции. Более того, в различных тканях (в мозге и печени, например) о нарушении работы митохондрий могут говорить различные маркеры, а изменение работы митохондрий происходит с разной скоростью. Поэтому очень трудно «перенести» данные, полученные для одного материала, на другой, и выбор оптимального материала для диагностики непрост.
- Проблема с подбором референсных значений. Определенные «границы нормы» значений для маркеров нарушения работы митохондрий разработаны только для митохондриальных заболеваний. «Норм» для возрастных изменений не существует. Для оценки степени выраженности возрастной дисфункции митохондрий необходим анализ больших баз данных.
Между тем, маркеры для оценки митохондриальной дисфункции существуют, и новые диагностические методики находятся в разработке.
Всех построим в ряд
Все существующие и потенциальные маркеры диагностики митохондриальной дисфункции можно подразделить на биохимические и генетические.
Биохимические маркеры
Эти маркеры косвенно свидетельствует о нарушении работы дыхательной цепи. Группа методик по определению биохимических маркеров описана в руководстве по диагностике митохондриальных заболеваний (определение уровня лактата, пирувата, их соотношения; количественное определение аминокислот, органических кислот и ацилкарнитинов; измерение количества ферментов — креатинкиназы, цитратсинтазы, сукцинатдегидрогеназы, цитрохром-с-оксидазы). Данные методы достаточно удобны, однако обладают невысокой специфичностью [46].
Также к биохимическим относят и более новые подходы: метаболомику, определение количества сывороточных цитокинов и микроРНК. Они используются для оценки вклада митохондриальной дисфункции при диабетической нефропатии [47], болезни Альцгеймера [48], биполярных нарушениях [49], для дифференциальной диагностики при сердечной недостаточности [50].
Что касается цитокинов, то в последние годы было установлено, что сывороточный фактор роста фибробластов 21 (FGF-21) и сывороточный фактор роста и дифференцировки 15 (GDF-15) — два многообещающих диагностических маркера митохондриальных заболеваний. Они более чувствительны и специфичны, чем используемые в клинической практике, но еще не включены в официальные протоколы. Эти факторы лучше использовать для диагностики митохондриальных дефектов трансляции и поддержания мтДНК, чем для патологий дыхательных комплексов или факторов сборки [51].
Генетические маркеры
Это очень важная группа маркеров, так как нарушение генетического аппарата митохондрий — не только следствие, но и причина митохондриальной «поломки». Дисфункция митохондрий сопровождается накоплением в них генетических патологий: как точечных мутаций, так и делеций длинных последовательностей ДНК. Количество генетических нарушений мтДНК растет с возрастом в мозге, сердце, толстом кишечнике, скелетной мускулатуре. Еще более неприятно то, что даже в самых юных клетках нашего организма — стволовых клетках — с возрастом увеличивается количество митохондрий с мутациями [52], [53].
К этой группе методов относят определение:
- Паттерна микроРНК. МикроРНК — некодирующие молекулы рибонуклеиновой кислоты длиной всего примерно в 20 нуклеотидов, которые контролируют генную экспрессию [54]. Сывороточные микроРНК всё чаще используются в целях диагностики, в том числе при патологиях, потенциально ассоциированных с дисфункцией митохондрий: заболеваниях мышц, митотической дистрофии, метаболических нарушениях. Было показано, что при митохондриальной энцефалопатии в клетках, несущих мутацию мтДНК m.3243 A>G, выявляется определенный паттерн микроРНК [55].
- Количества копий внутриклеточной мтДНК. В статье [56] использовали линию мышей со встроенной генной конструкцией, которая позволяла «включать» истощение мтДНК и «выключать» его, восстанавливая мтДНК. У мышей при истощении пула мтДНК наблюдались внешние признаки старения — морщинистость кожи и облысение. При этом восстановление мтДНК полностью обращало вспять данные изменения.
Количество копий мтДНК в Т-клетках, по всей видимости, связано со старением иммунной системы. Дело в том, что Т-клетки обычно находятся в спящем состоянии, пока им не дадут наводку на тот или иной патоген. Тогда их метаболизм существенно меняется. В том числе, происходит увеличение количества копий мтДНК. Но, как оказалось, у пожилых людей копирование мтДНК после активации Т-клеток происходит в гораздо меньшей степени, чем у молодых. При этом Т-клетки долгожителей сохраняют способность к быстрому увеличению количества копий мтДНК при вторжении патогена. Это было показано в работе, сравнивающей иммунный ответ у молодых и больных/здоровых долгожителей [57].
А еще недавно было обнаружено, что восстановление должного количества копий мтДНК замедляет старение сосудов у мышей. Это было определено по показателям снижения эластичности сонной артерии, скорости пульсовой волны в аорте и содержанию в ней эластина и коллагена [58].
Помимо точечных мутаций к генетическим нарушениям митохондрий относятся делеции — выпадение относительно крупных фрагментов митохондриального генома. Известно около 100 делеций мтДНК, которые, вероятно, участвуют в процессе старения. Самая частая соматическая мутация, которая накапливается при старении в мтДНК, — делеция 4977-bp [57], [59] (ее так и называют — «частая делеция»). - Количества внеклеточной мтДНК (вкмтДНК). вкмтДНК циркулирует в крови либо в свободном виде, либо в составе внеклеточных везикул. И хотя основная функция вкмтДНК — сигнальная, высокий уровень вкмтДНК ассоциирован с хроническими заболеваниями и используется в качестве их биомаркера [60], [61].
- Уровня гетероплазмии по точечным мутациям и «частой делеции» 4977-bp. Ее уровень с возрастом растет, а митохондрии с такой делецией не могут функционировать. Ее накопление происходит в тканях с низкой пролиферативной активностью (скелетные мышцы, мозг, сердце). Криминалисты используют этот маркер в своей работе для определения возраста [62]. Делеция обнаруживается в клетках кожи при фотостарении и даже в волосах.
- Уровня метилирования мтДНК — ключевого эпигенетического процесса, регулирующего экспрессию генов, в том числе митохондриальных. Всё больше данных свидетельствует в пользу того, что уровень метилирования ДНК митохондрий связан с окислительным стрессом и старением клетки [63], [64].
А ну-ка глянем, что там?
К методам визуальной оценки относят как инвазивные методики биопсии с последующей оценкой объемной плотности митохондрий, так и неинвазивные — магнитно-резонансную спектроскопию и позитронно-эмиссионную томографию.
Небольшой укольчик
Биопсия инвазивна и дорогостояща, но она всё еще один из основных методов диагностики митохондриальных нарушений. Чаще всего проводят биопсию скелетных мышц или кожи. При подозрении на заболевание также возможна биопсия печени, миокарда. Взятый образец можно анализировать гистохимическими и иммунологическими методами, оценить количество митохондрий в образце ткани, их интактность. Кроме этого, можно выделить митохондрии и провести функциональную оценку (изучить ультраструктуру и сделать биохимический анализ дыхательной цепи).
Волшебный магнит
Магнитно-резонансная терапия (МРС) — метод исследования химических веществ с использованием явления магнитного резонанса. МРС обычно применяется в качестве инструмента для дифференциальной диагностики поражений головного мозга, но может использоваться и для изучения миопатий. Этот метод позволяет проводить количественную оценку метаболитов тканей в устойчивом состоянии. Для оценки работы митохондрий чаще всего используются спектры 31P (изотопа фосфора) и 1H (водорода). 1H позволяет помечать тканеспецифичные метаболиты, такие как лактат, холин и N-ацетиласпартат, и оценивать их содержание в тканях. В то же время использование 31P позволяет оценить относительные пропорции метаболитов фосфора и, следовательно, окислительную емкость. 31P-МРС в скелетных мышцах позволяет идентифицировать ключевые различия в биоэнергетике тканей при различных нейромышечных нарушениях [65].
И позитроны тоже!
В отличие от МРС, которая обеспечивает измерение метаболитов в стабильном состоянии, позитронно-эмиссионная томография (ПЭТ) измеряет метаболический поток, что позволяет изучать метаболические и гемодинамические свойства тканей [66]. Для изучения дисфункции митохондрий используются радиоизотопно-меченные метаболиты 15O, 2-фтор-18F-2-дезокси-D-глюкоза и 11C-пируват, которые характеризуют тканеспецифичную энергетику. При помощи этого метода наиболее изучены пациенты с MELAS-синдромом — прогрессирующим нейродегенеративным заболеванием, характеризующимся энцефалопатией, лактацидозом и инсультоподобными эпизодами. С помощью этой методики у таких больных изучали ткани сердца, мышц и мозга [51], [68].
Спортивные мероприятия
Метод функциональной диагностики — эргоспирометрия — часто используется спортсменами. Для этого проводят нагрузочный тест (человек крутит педали велотренажера или бежит по беговой дорожке) и анализируют газообмен в состоянии покоя, во время нагрузки и в период восстановления. При этом измеряют потребление кислорода (VО2), выдыхаемый углекислый газ (VСО2) и вентиляционные параметры, иногда добавляют биомаркеры в крови, в основном лактат. Нарушения митохондриальной функции снижают аэробный потенциал, что фиксируется эргоспирометрией и лактат-стресс-тестом [69], [70].
Метод может использоваться как для подтверждения дисфункции митохондрий, так и для оценки эффективности терапии. Он неинвазивный, доступный и достаточно чувствительный, хоть и требует специального оборудования и подходит не для всех возрастных групп [71].
Лечение митохондриальных недугов
Консервативная терапия
Физическая активность — наиболее доказанный способ оптимизации митохондриальной функции. Регулярные тренировки в течение целого месяца увеличивают содержание митохондрий в клетках на 30–100%, а их объемную плотность на 40%. Если заниматься полгода, активируется системный биогенез митохондрий, предотвращается истощение пула мтДНК, снижается скорость образования мутаций в мтДНК, увеличивается митохондриальная окислительная емкость, а кристы делаются более «компактно упакованными», за счет чего оптимизируется работа дыхательной цепи митохондрий [72]. Биогенез митохондрий повышает максимальное потребление кислорода, оптимизирует процессы поглощения кислорода для окислительного фосфорилирования и окисления жирных кислот (на единицу потребленного субстрата образуется больше энергии АТФ, которую клетка может использовать). Кроме этого, если окисление жирных кислот происходит частично, то накапливаются промежуточные продукты (церамиды и диацилглицеролы). Сокращение мышцы запускает ряд изменений (повышение уровня внутриклеточного кальция, снижение значения АТФ/АМФ, повышение значения НАД+/НАДH и повышение количества АФК), влияющих на основные внутриклеточные сигнальные пути, которые контролируют работу митохондрий (АМФ-активируемую протеинкиназу, фактор, индуцируемый гипоксией 1-альфа), и на синтез PGC1ɑ — главного регулятора образования митохондрий [73].
При этом как длительные тренировки умеренной интенсивности, так и высокоинтенсивные интервальные тренировки и даже интервальный спринт (который считается типично анаэробной историей и занимает 10 мин в неделю) запускают сходное изменение в уровнях маркеров биогенеза митохондрий: повышение количества цитратсинтазы и цитохром-С-оксидазы. Но только длительные тренировки приводят к росту количества митохондрий, а кратковременные интервальные лишь влияют на эффективность деятельности уже существующих, меняя их ультраструктуру и оптимизируя работу дыхательной цепи [73].
Про влияние питания на работу митохондрий в интернете можно найти массу информации, более или менее достоверной. Самое важное — переключиться со сжигания углеводов на жиры. Этого можно достигнуть, в первую очередь, за счет периода ночного голода не меньше 12 часов. Переедание и вызванная им «перегрузка» работой (ведь всю эту еду, в конечном счете, должны окислить митохондрии) однозначно нарушает работу митохондрий. Короткие периоды голода стимулируют динамику митохондрий , положительно влияя на системы контроля качества и работу дыхательной цепи.
Митохондрии непрерывно находятся в динамике, регулируя поступление или расход энергии. Ведь мы «кормим» свои клетки или слишком много, или слишком мало, а в ответ требуем выдать то количество АТФ, которое точно должно соответствовать нашим потребностям. Чтобы регулярно выходить из этой ситуации, митохондрии и используют своеобразные «движения» — деление и слияние между собой. Эти «митодвижения» объединяют под названием «динамика митохондрий». Баланс между делением и слиянием митохондрий — центральный механизм биоэнергетической адаптации к метаболическим потребностям клетки. Так, если в клетку какой-либо из тканей (кроме некоторых нейронов в мозге) поступает большое количество питательных веществ, и поступление превышает затраты, то митохондрии находятся во фрагментированном состоянии. Если клетка голодает, и поступления меньше затрат, митохондрии сливаются.
В чем смысл этих движений? Если клетка голодает, то слияние митохондрий позволяет увеличить их биоэнергетическую эффективность. Если же еды избыток, то задача митохондрий — рассеять побольше энергии в виде тепла, чтобы избежать окислительного стресса. Фрагментация митохондрий позволяет им снизить биоэнергетическую эффективность и защитить клетку от окислительного стресса. Кроме этого, нормальный цикл деления митохондрий и их слияния — это ключевое звено контроля их качества [74–80].
Что касается витаминов, то для нормальной работы дыхательной цепи необходимо адекватное (а не избыточное!) поступление в организм цинка, магния, витаминов группы В, витаминов С и Е. При построении плана правильного питания это обязательно нужно учитывать.
На работу митохондрий также влияет ряд компонентов, входящих в БАДы (коэнзим Q, карнитин, нитраты, кофеин, альфа-липоевая кислота, таурин, мелатонин). Их эффективность подтверждена далеко не во всех случаях и сильно зависит от возрастной группы и степени дисфункции митохондрий.
Питание также может оказывать свое воздействие на митохондрии через микробиом. Да данный момент расшифровано более трех миллионов генов микробного сообщества — это в 150 раз больше, чем полное число генов в нашем геноме [81]. Микроорганизмы, населяющие наш кишечник, выделяют разнообразные метаболиты, которые влияют на наш организм и, в частности, на митохондрии. Возможно, способность так чутко реагировать на метаболиты микроорганизмов осталась у митохондрий со времен «бактериального прошлого». Взаимодействие микробиом—митохондрии сейчас в топе научных интересов не только с теоретической точки зрения. Используя знания из этой области, можно разработать способ повлиять на наши митохондрии при помощи модификации микробиома. Сделать это вполне реально: в определенных пределах использование тех или иных пищевых компонентов, пре- и пробиотиков может изменить количество микроорганизмов, выделяющих те или иные метаболиты [82–84].
Метаболиты микробиома можно разделить на группы по типу их воздействия на митохондрии:
- Оксид азота (NO) ингибирует цикл трикарбоновых кислот, снижая образование ацетил-КоА (основного энергетического субстрата) в митохондриях.
- Сероводород (H2S) в высоких концентрациях ингибирует комплекс IV дыхательной цепи. В низких — способствует митофагии и повышает биогенез [85].
- Короткоцепочечные жирные кислоты (ацетат, пропионат и бутират). Все они, особенно бутират, являются «топливом» для дыхательной цепи митохондрий.
- Уролитин А — метаболит, образуемый некоторыми бактериями кишечника из эллаговой кислоты (содержится в гранатах, малине, орехах). Потребление уролитина А пожилыми грызунами способствовало росту выносливости и силы мышц без увеличения их массы и приводило к увеличению количества митохондрий. Кроме того, на культуре клеток было показано, что использование уролитина А запускало митофагию [86]. Сейчас проходят клинические испытания (NCT03464500 и NCT03283462) на здоровых пожилых добровольцах, которые уже показали безопасность его применения [85].
Может быть, стоит оказать на них влияние?
Значительная часть препаратов — потенциальных геропротекторов — оказывает воздействие на митохондрии. Рассмотрим некоторые из них.
- АИКАР (5-аминоимидазол-4-карбоксамид рибофуранозид) — потенциальный миметик физической активности, запрещен WADA (World Anti-Doping Agency, Всемирное антидопинговое агентство) как допинг. АИКАР — один из предшественников пуриновых нуклеотидов, то есть, по сути, природное соединение; поэтому его нельзя запатентовать как лекарственный препарат. АИКАР продается в качестве нутрицевтика, например компанией Peptide Sciences(США).
АИКАР, будучи введенным в организм млекопитающего, быстро превращается в АИКАР-фосфат — природный аналог аденозинмонофосфата. Накопление АИКАР-фосфата в организме сигнализирует о недостатке АТФ, или, проще говоря, о дефиците энергии. Сигнал АИКАР-фосфата или АМФ воспринимается ферментом АМФ-активируемой протеинкиназой (АМФК), фиксирующей дефицит энергии и блокирующей процессы, связанные с потреблением АТФ, стимулируя при этом синтез АТФ и биогенез митохондрий. - Аспирин, употребляемый в небольших дозах, приводит к разобщению дыхательной цепи в митохондриях, что снижает образование активных форм кислорода, но, в то же время, может приводить к апоптозу клеток слизистой желудка [88]. Аспирин также вызывает образование новых митохондрий [89], [90].
- Метформин. Данные по влиянию метформина на функции митохондрий довольно противоречивы. С одной стороны, он стимулирует АМФК, что должно усиливать биогенез митохондрий, с другой — снижает уровень синтеза АТФ, разобщает дыхательную цепь, приводит к истощению пула коэнзима Q. Метформин может снижать уровни физических показателей [91], но в то же время не исключено и положительное воздействие легкого разобщения дыхательной цепи у людей старшей возрастной группы. Так, показано, что метформин улучшал работу митохондрий, снижая уровень системного стерильного воспаления [92], хотя даже в группе пожилых пациентов метформин снижает показатели адаптации к спортивным нагрузкам [93].
- Ресвератрол стимулирует биогенез митохондрий и оптимизирует показатели работы сердечно-сосудистой системы и метаболизм скелетных мышц у лиц, страдающих ожирением. А вот у спортсменов он понижает уровни показателей адаптации к физическим нагрузкам, предположительно за счет антиоксидантного воздействия [94].
Мишень — кардиолипин
Кардиолипин — фосфолипид, который в норме находится только во внутренней мембране митохондрий и там же синтезируется. Он состоит из четырех ацильных цепочек, которые «удерживаются» глицерином и двумя фосфатными головками (рис. 10).

Рисунок 10. Химическая формула кардиолипина
Жирные кислоты, входящие в состав кардиолипина, различаются от ткани к ткани. В митохондриях мышц и сердца это в основном линолевая кислота. В мозге кардиолипин имеет более разнообразный жирнокислотный состав: он содержит полиненасыщенные жирные кислоты, арахидоновую и докозагексаеновую кислоты. Кардиолипин имеет уникальную коническую форму, которая агрегирует в небислойные структуры и способствует образованию крист митохондрий. Благодаря кардиолипину формируется пространственная структура внутренней мембраны, в которой комплексы дыхательной цепи сильно сближаются, формируя так называемые суперкомплексы. Благодаря этому электроны переходят от комплекса к комплексу быстрее, их утечка и образование АФК снижаются, а скорость синтеза АТФ растет.
Дефицит кардиолипина ведет к нарушению структуры крист и снижению синтеза АТФ. Ведь если связь между комплексами нарушается, и они расположены не оптимальным образом, то образование свободных радикалов усиливается, а синтез АТФ падает.
В кардиолипин входят ненасыщенные жирные кислоты, подверженные окислению. Для митохондрий это плохо, так как полупериод жизни кардиолипина гораздо больше, чем у других фосфолипидов, и он реже обновляется. Старение сопровождается истощением пула кардиолипина и окислительным стрессом [95]. Но самое интересное не то, что кардиолипина становится меньше, а то, что он становится другим. При старении происходит его ремоделирование, линолевой кислоты становится меньше, а полиненасыщенных кислот — больше. Нарушается поток электронов по дыхательной цепи, их утечка растет, повышается выработка свободных радикалов, а энергии образуется меньше. Но еще неприятнее, что ремоделирование кардиолипина нарушает митофагию (рис. 11) [87]. При повреждении митохондрий кардиолипин перераспределяется на внешнюю митохондриальную мембрану и «флажком» маркирует «поломанные» митохондрии, которые быстро удаляются при помощи митофагии. Однако окисление кардиолипина приводит к высвобождению цитохрома С из митохондрий и последующему апоптозу клетки. Вообще следует сказать, что организм плохо воспринимает кардиолипин вне митохондрий, и запускает стерильное воспаление, так как эта молекула очень похожа на фрагмент бактерии.

Рисунок 11. Биологические функции кардиолипина: участие в сборке и функционировании дыхательных суперкомплексов и переносе через них субстрата и электронов; участие в динамике митохондрий; повышение митохондриального биогенеза белков; участие в образовании крист митохондрий. Кроме того, кардиолипин — платформа для интегрирования процессов контроля качества митохондрий.
Неудивительно, что существенная роль кардиолипина в митохондриальных процессах и изменение его структуры с возрастом делают его интересной целью митохондриальной медицины. Подходы тут самые разные.
Например, для терапии могут использоваться фосфолипиды — прекурсоры кардиолипина. Один из них — фосфатидилглицерол. В исследовании in vitro было показано, что фосфатидилглицерол встраивается в мембраны митохондрий, при чем повышается уровень синтеза АТФ и растет потребление кислорода, а также снижается продукция провоспалительных цитокинов [97]. В экспериментах in vivo всё, конечно, намного сложнее, так как возникают вопросы о биодоступности исследовательского материала.
Помимо фосфолипидов-предшественников, есть еще коммерческий фармакологический препарат эламипретид. Он поглощается клетками при помощи пассивной диффузии, проникая даже в деполяризованные митохондрии, и взаимодействует с кардиолипином. In vitro было показано, что эламипретид оказывает на организм следующее воздействие:
- снижает образование АФК;
- повышает уровень поглощения кислорода;
- снижает окисление кардиолипина;
- повышает количество АТФ [98], [99].
У мышей эламипретид повышал выносливость и ремоделировал структуру крист митохондрий в канальцах почек при метаболическом синдроме, в эндотелии сосудов при ишемии, в сетчатке глаза при диабетической ретинопатии. Возможно, он даже способствует сохранению жирнокислотного состава кардиолипина и восстанавливает динамику митохондрий и митофагию [99–101].
Исследований по этому пептиду накоплено уже очень много, и на грызунах он показывает довольно многообещающие результаты. Например, старые мыши бегали почти как молодые после восьми дней терапии этим препаратом, причем положительные изменения начались уже через час после введения [102]! Это говорит об улучшении митохондриальной функции именно за счет защиты кардиолипина от структурных изменений и улучшения качества работы ЭТЦ. Также он повышает когнитивные способности у старых мышей, при этом положительно влияя на эндотелий сосудов головного мозга [103]. У здоровых пожилых людей уже после однократного введения эламипретид улучшал показатели мышц до уровня, сопоставимого чуть ли не с уровнем, которого можно достигнуть в течение полугода тренировок.
Эламипретид сейчас проходит клинические испытания второй фазы (NCT02805790, NCT02693119, NCT02976038). Недавно две компании: Stealth BioTherapeutics (специализация — митохондриальные заболевания) и Alexion Pharmaceuticals (специализация — редкие заболевания) подписали соглашение для совместной разработки и коммерциализации этого препарата.
Скажи «нет» окислению!
Если раньше, согласно теории Хармана, АФК считали главной причиной старения и всевозможных повреждений, то сейчас мы знаем, что это не совсем так.
Сегодня известно, что АФК — это не столько «зло», сколько сигнал к действию. И только когда организм не может отреагировать на этот сигнал должным образом (усилив антиоксидантную защиту, устранив поврежденные митохондрии, обеспечив работающие митохондрии недостающими им белками) — только тогда свободные радикалы становятся орудием массового поражения, что приводит к окислительному стрессу, воспалению и старению. У здоровых людей использование антиоксидантов может снижать показатели адаптации к спортивным нагрузкам. Поэтому их эффективность для улучшения состояния организма в значительной мере зависит от выбранной целевой группы.
Для нейтрализации АФК в организме существуют различные системы антиоксидантной защиты (эндогенные антиоксиданты). Но как бы ни совершенна была наша клеточная система антиоксидантной защиты, иногда ее оказывается недостаточно для предотвращения развития окислительного стресса и воспаления. Тогда возможно использование экзогенных антиоксидантов. Несмотря на то, что свободнорадикальная теория старения была пересмотрена, поиск новых антиоксидантов и оптимизация их работы всё еще находятся в топе биотехнологических исследований в митохондриальной медицине, и антиоксиданты представляют собой наиболее обширную группу препаратов, используемых при терапии дисфункции митохондрий.
Необходимо отметить, что понятие «антиоксиданты» очень широкое. К ним относятся совершенно разные соединения и молекулы, которые способны:
- усиливать естественные системы антиоксидантной защиты;
- снижать утечку электронов и уровень образования АФК;
- непосредственно нейтрализовать АФК, связываясь с ними.
Антиоксиданты могут снижать окислительный стресс, но они не влияют на производство АТФ и работу дыхательной цепи. Они могут действовать как в цитоплазме (например витамин С), так и в липидной фракции мембран (витамин Е). Так как митохондрии в клетке являются основным местом образования АФК, то разрабатываются также таргетные антиоксиданты, действие которых направлено непосредственно на митохондрии.
К группе таргетных антиоксидантов относятся SkQ (ионы Скулачева) и MitoQ. Они могут проходить через двойную мембрану митохондрий, воздействуя непосредственно в месте образования свободных радикалов — вблизи ЭТЦ. Молекула SkQ состоит из двух частей: челнока, который протаскивает ее через мембрану митохондрий, и антиоксиданта пластохинона (растительного аналога убихинона, или коэнзима Q10).
Препарат MitoQ по своему механизму действия очень сходен с SkQ. Молекула митохинона (MitoQ) состоит из переносчика через митохондриальную мембрану и убихинона. Однако оба эти соединения не проникают в деполяризованные митохондрии, так как их способность проходить через митохондриальную мембрану напрямую зависит от ее заряда.
На искусственных моделях старения MitoQ показывает стандартно хорошие результаты. Выяснено, что использование препарата улучшает сосудистую функцию у пожилых людей [104]. Лекарственный препарат с SkQ «Визомитин» (глазные капли) нацелен на различные возрастзависимые офтальмологические проблемы — синдром сухого глаза (3 фаза клинических испытаний), увеиты (2 фаза), сухая макулярная дегенерация (2 фаза). А препарат «Пластомитин» — на рассеянный склероз, острое повреждение почек, синдром Барта, нейродегенеративные заболевания.
Другим интересным классом соединений, которые оказывают антиоксидантное воздействие, являются широко применяемые препараты для сердца. Среди них ингибиторы ренин-ангиотензиновой системы, ангиотензин-превращающего фермента (АПФ) (эналаприл, рамиприл), а также сартаны (блокаторы рецептора ангиотензина 1 типа) [105].
Ренин-ангиотензиновая система поддерживает в организме водно-солевой баланс, регулируя таким образом артериальное давление, вазоконстрикцию, накопление клетками натрия и выведение из них калия. При ожирении и старении происходит повышение активности системы РААС, чему отводится большая роль в развитии возрастзависимых патологий: росту стерильного воспаления, развитию дисфункции эндотелия, фиброзу, гипертонии, атеросклерозу, возрастному ремоделированию тканей сердца, болезням почек [106].
Главный эффект ангиотензина — сужение сосудов. Так как это влияет на количество кислорода, то логично предположить, что митохондрии как основные потребители кислорода должны как-то реагировать на изменение количества ангиотензина. И правда, недавно обнаружили, что у митохондрий есть два класса рецепторов к ангиотензину — АТ1 и АТ2. При этом в зависимости от того, с каким из рецепторов свяжется ангиотензин, действие его разнится. Активация АТ2 приводит к снижению митохондриального дыхания и повышению концентрации оксида азота. Эти эффекты защищают от окислительного стресса и вызванного им повреждения. А вот активация АТ1, наоборот, приводит к образованию супероксида и окислительному стрессу. Уровень производства «хороших» рецепторов АТ2 существенно выше, а окислительный стресс приводит к дополнительному компенсаторному усилению их активности.
Однако старение влияет на синтез рецепторов ангиотензина. Казалось бы, должен происходить компенсаторный рост выработки «хороших» АТ2 рецепторов, которые бы снижали окислительный стресс. Однако наблюдается прямо противоположная ситуация: производство АТ2 падает при старении, а вот АТ1, приводящих к образованию супероксид-аниона, растет [107]. Поэтому получается «двойной оксидативный удар» — рост активности РААС и синтеза рецепторов АТ2. Именно поэтому ингибиторы АПФ (эналаприл, рамиприл), снижающие образование ангиотензина, и блокаторы рецептора ангиотензина первого типа (сартаны) оказывают антиоксидантное воздействие.
А если подправить гены?
В то время как методы генной терапии мутаций ядерной ДНК развиваются довольно активно (особенно для моногенных заболеваний) [108], [109], с митохондриями всё гораздо сложнее. Дисфункция наступает только тогда, когда количество митохондрий с мутацией преодолевает некоторую пороговую черту. Мы не можем исправить мутацию во всех митохондриях — в клетке их слишком много. Бóльшая часть методов генной терапии направлена на изменение соотношения митохондриальной гетероплазмии (мутантные → нормальные митохондрии). Общие принципы такой терапии изображены на рисунке 12.

Рисунок 12. Принцип действия генной терапии митохондрий: направленное повреждение мутантных копий мтДНК с последующей репликацией здоровых копий
Именно поэтому практически все методы генной терапии митохондрий направлены не на коррекцию мутантных генов, а на прицельную деградацию ДНК в митохондриях с мутацией, что изменяет соотношение гетероплазмии в сторону немутантных. Данное направление генной терапии носит название «антигеномное», или «антирепликативное».
В нем различают несколько методических подходов, которые мы коротко опишем с указанием их основных преимуществ и недостатков.
Использование ДНК-нуклеаз
Этот метод предполагает использование генно-инженерных конструктов, которые кодируют сайт-специфичные ДНК-нуклеазы (ферменты, расщепляющие нуклеиновые кислоты строго в определенной точке), нацеленные на ДНК митохондрий. Их можно использовать для прицельного удаления последовательности мтДНК с мутацией, что позволит сдвинуть гетероплазмию в сторону здоровых митохондрий путем внесения в ядерный геном последовательности, кодирующей нужный фермент [111].
Механизм действия таков: если в митохондриальном геноме возникает мутация и появляется специфическая нуклеотидная последовательность, то фермент разрезает ДНК, и такая митохондрия с большой вероятностью будет ликвидирована. Подобные опыты проводились на культурах гепатоцитов мыши: использование генной терапии позволяло изменить уровень гетероплазмии за шесть часов. In vivo такой метод проверяли на мышиной модели при помощи внесения аденовирусного вектора. При этом эндонуклеазы рестрикции, кодируемые им, эффективно изменяли уровень гетероплазмии в сторону здоровых митохондрий. Этот метод довольно безопасен — у подопытных мышей не происходило снижения общего уровня мтДНК и не возникало дополнительных проблем со здоровьем [112], [113].
Недостаток этого метода — сложность в поиске той последовательности-мишени, которая будет присутствовать только в мутантных митохондриях, но не в здоровых. Для более чем двухсот мутаций мтДНК, ассоциированных с заболеваниями, только у двух была обнаружена та единственная и неповторимая последовательность ДНК, подходящая в качестве мишени для нуклеаз.
Создание Zn-finger ДНК-связывающихся модулей (mitoZFN)
Инженерные «цинковые пальцы» (zinc finger nucleases, ZFNs) — это химерные ферменты, содержащие две структурные единицы: связывающуюся со строго определенной последовательностью ДНК и расщепяющую ее. Этот метод широко использовался и для редактирования ядерных геномов, но был вытеснен CRISPR-Cas-технологией. А вот для митохондриального геномного редактирования он по-прежнему применяется на клеточных и животных моделях митохондриальных заболеваний.

Рисунок 13. Генная терапия митохондрий при помощи «цинковых пальцев», разрушающих мутантные копии митохондриальной ДНК, снижая гетероплазмию
Такие модули «цинковых пальцев» были созданы для практически всех шестидесяти четырех нуклеотидных комбинаций кодонов. Для удобства распознавания и последующего удаления к такому модулю можно добавить специфический «флажок», облегчающий распознавание нужной последовательности — метильную метку. В данном случае это может делать метилтрансфераза DNMT3. Такой нуклеотид будет гораздо проще распознать. Благодаря этому комбинация (модуль mitoZFN, направленный на какую-то последовательность, плюс генный конструкт со специфической нуклеазой) позволяет таргетно убирать специфические последовательности мтДНК.
Интересно, что синтез нескольких комбинаций специфичных mtZFNs-модулей использовалась также для удаления «частой делеции» 4977-bp (о которой мы говорили выше) на модели цитоплазматических гибридов клеток. Данная делеция ассоциирована с офтальмоплегией и накапливается у всех людей в процессе старения в различных тканях.
Сложность данного метода в том, что большое количество модулей внутрь митохондрии доставить достаточно сложно.
mitoTALENs (transcription activator-like effector nucleases)
Более перспективным средством избирательного воздействия на мтДНК могут быть векторные конструкции на основе химерных нуклеаз, названные TALENs (transcription activator-like effector nucleases) [114]. Роль ДНК-распознающих структур в них играют белковые домены, каждый из которых распознает только один нуклеотид, природный прототип которых — белки некоторых бактерий, паразитирующих на клетках сельскохозяйственных растений [115].
При этом подходе нуклеазы распознают отдельные нуклеотиды и обеспечивают связывание и расщепление нуклеотидной последовательности, если она начинается с тимидина. Этот подход успешно применялся для изменения уровня гетероплазмии, например, в ооцитах мыши [116].
Такой же подход использовался на мышиной модели гетероплазматической митохондриальной мутации. Для этого была создана модель с мутацией митохондриального гена тРНК аланина. Эта мутация приводила к нестабильности этой тРНК и возникновению сердечно-сосудистых патологий у мышей в более старшем возрасте. В этом исследовании TALEN вводили внутримышечно, внутривенно и внутрибрюшинно. При этом в мышцах и сердце уровень мутантной мтДНК существенно снижался, и эффект был стабилен. Молекулярный дефект был успешно исправлен таким лечением [117].
Основной недостаток этого метода — сложность доставки большого количества TALEN внутрь митохондрии, так как вектор может быть слишком большой.
Редактирование генома при помощи системы CRISPR-Cas9
CRISPR-Cas9 — технология редактирования геномов высших организмов, базирующаяся на иммунной системе бактерий [118]. Данная методика, которая получила широкое распространение для редактирования ядерного генома и совершила революцию в этой области [119], всё еще имеет ограничения для редактирования митохондриального генома. Принцип работы «генетических ножниц» CRISPR-Cas9 — направленное разрезание ДНК в избранных участках. Для этого искусственно синтезируют CRISPR-РНК, в которую включают последовательность, выбранную исследователем. Белок Cas9 способен распознать и связаться с такой синтетической СRISPR-РНК (ее называют «гид», или «направляющая РНК») и разрезает соответствующее место в ДНК [120].
Что касается редактирования митохондриального генома, то тут возникают сложности, так как, во-первых, довольно сложно доставить направляющую РНК внутрь митохондрии, что необходимо для нацеливания белка Cas9. Во-вторых, в митохондриях низкий уровень репарационных процессов, отвечающих за восстановление двунитевых разрывов. При редактировании же генома результат работы CRISPR-Cas9 — это починка двухцепочечного разрыва, произведённого Cas9. С низким уровнем репарации этот процесс протекает довольно сложно.
Общий недостаток всей этой группы методов генного редактирования (а по сути — удаления копий мтДНК, несущих мутацию) — возможность снизить общее количество мтДНК ниже функционального порогового уровня. Это усугубляет проблему митохондриальной дисфункции и может привести к апоптозу клетки.
Так как же вылечить митохондрии?
Сегодня используется в основном консервативное лечение митохондриальных заболеваний, которые не помогают, если степень митохондриальной дисфункции высока или если есть врожденные митохондриальные заболевания. Нормальная работа митохондрий нарушается не только при митохондриальных заболеваниях, но также при болезнях Альцгеймера и Паркинсона, диабете, саркопении, инсулин-резистентности и при таких состояниях как инсульт, инфаркт миокарда, а еще в процессе старения организма. При этом известно, что доставка новых митохондрий или восстановление их пула может давать терапевтический эффект.
По словам ученых, новая парадигма митохондриальной терапии основана на доставке «свежих» органелл [121] — как аутологичных (из клеток того же организма), так и гетерологичных (из других организмов). Искусственное внесение в клетку митохондрий может решить главную проблему возрастной митохондриальной дисфункции: увеличить количество «хороших» митохондрий до порогового уровня, необходимого для нормальной работы клетки.
Необходимо заметить, что in vivo передача митохондрий от клетки к клетке — это нормальный и довольно распространенный в организме процесс. Клетки в нашем организме могут обмениваться мтДНК (в составе экзосом, микровезикул, апоптотических телец) и даже передавать другим клеткам целые митохондрии (при помощи микровезикул и туннельных нанотрубочек). Донорами митохондрий могут быть мезенхимальные стволовые клетки, фибробласты, астроциты, активированные тромбоциты и некоторые другие типы клеток [122].
Перенос митохондрий in vivo выполняет целый ряд физиологических функций:
- спасение клетки от апоптоза при ее повреждении (так, например, астроциты «спасают» нейроны) [123];
- перепрограммирование клеток и их дифференцировка [124], [125];
- участие в иммунных процессах (трансфер митохондрий от мезенхимальных стволовых клеток к макрофагам усиливает фагоцитарную активность последних) [126].
Что касается искусственного переноса митохондрий, тут можно выделить два основных подхода.
Митохондриальная обогащающая терапия
Это «трансплантация» митохондрий (аутологичных или гетерологичных) in vitro с последующим внедрением «обогащенных» клеток обратно в организм (рис. 14) [127].

Рисунок 14. Принцип проведения митохондриальной обогащающей терапии
Терапия обогащения митохондриями ооцитов при бесплодии использовалась довольно широко, при этом для введения в организм-реципиент брали донорские митохондрии [128–130]. В ооцит, полученный от женщины с проблемами зачатия, вводили фрагмент цитоплазмы с митохондриями из клеток молодой женщины без таковых. Результаты были положительные, но в 2005 году FDA (Food and Drug Administration) запретила этот метод, поскольку был недостаточно изучен риск митохондриальной гетероплазмии. Сейчас терапия обогащения митохондриями используется с применением аутологичных митохондрий, полученных из предшественников половых клеток самого пациента (NCT02586298).
Для лечения митохондриальных заболеваний данный подход запатентовала и использует компания Minovia (Израиль). Пока что в исследование вовлечено пять пациентов с синдромом Пирсона.
Трансфер выделенных изолированных митохондрий непосредственно в ткань или системно в кровоток
Большая часть исследований этого метода проводилась на крысах и кроликах. Трансфер митохондрий на животных моделях успешно использовали для лечения ишемии сердца, постинсультных состояний, острого повреждения легких. При этом брали как аутологичные митохондрии, так и митохондрии от других особей и даже видов [131].
Митохондриальная терапия обещает успешные результаты при различных патологиях: ишемии миокарда, болезни Паркинсона, острой печеночной недостаточности [132–134].
Врачи, работающие над митохондриальной терапией, утверждают, что в ближайшее время эта технология может быть внедрена в качестве стандартного терапевтического подхода в случае внезапного массивного повреждения тканевых митохондрий, в частности, в случаях ишемии сердечной мышцы [133–135].
В ходе развитие митохондриальной терапии потребуется решить еще немало вопросов. Например, очень важно оценить влияние системного и локального введения митохондрий на биомаркеры старения в животных моделях. Важно изучить, будет ли нормально реплицироваться ДНК пересаженных митохондрий, будут ли они способны к нормальному делению и другим процессам?
Ну и конечно, остается совершенствовать методологию. Очень нужно стандартизировать процедуры изоляции митохондрий, подобрать оптимальные концентрации для инъекции, тестировать среды, в которых находятся органеллы, и разработать методики оценки качества и целостности митохондрий. А еще важно выбрать оптимальный источник клеток и тканей для получения митохондрий. На данный момент, к сожалению, единого мнения насчет того, какой источник лучше всего, нет.
Заключение
Митохондрии — удивительные механизмы, работающие на благо нашего здоровья. Они придают нам энергию, защищают от патогенов, участвуют в дыхании. Совсем не удивительно, что ученым пришло в голову использовать их в качестве мишени для терапии различных, в том числе возрастных, заболеваний. Ведь болеет митохондрия — нарушаются многие важнейшие процессы в организме.
Митохондриальная медицина развивается стремительно, направлений и идей в ней множество и каждый день их арсенал пополняется. Митохондриальная медицина — это и лечение, и диагностика. Показатели здоровья митохондрий используются для оценки состояния организма с помощью довольно широкого арсенала методов: определение генетических и биохимических маркеров патологий митохондрий, визуальные методики, МРС, ПЭТ, функциональная диагностика. Простора для фантазии не занимать!
Направлений, в которых нужно работать — множество. Это и консервативная терапия, и генное редактирование, и использование антиоксидантов. Действовать надо всем вместе и сообща! Разработки появляются стремительно, и, кто знает, возможно, скоро они приведут нас к успеху в победе над старением?
Литература
- Опасные связи. Новый взгляд на происхождение эукариотических химер, подмявших под себя весь мир;
- Внимание! Разыскивается предок митохондрий!;
- Eugene V. Koonin. (2015). Archaeal ancestors of eukaryotes: not so elusive any more. BMC Biol. 13;
- Purificación López-García, Laura Eme, David Moreira. (2017). Symbiosis in eukaryotic evolution. Journal of Theoretical Biology. 434, 20-33;
- Purificación López-García, David Moreira. (2015). Open Questions on the Origin of Eukaryotes. Trends in Ecology & Evolution. 30, 697-708;
- Вторая жизнь АТФ: от главной батарейки до нейромедиатора;
- Особая диета: молекулярный водород три раза в день, ионы сульфата перед едой;
- Denham Harman. (1992). Free radical theory of aging. Mutation Research/DNAging. 275, 257-266;
- Нобелевская премия по медицине и физиологии 2016: за самоедство;
- Аутофагия, протофагия и остальные;
- Navdeep S Chandel. (2014). Mitochondria as signaling organelles. BMC Biol. 12;
- Война и мир: как устроить белковую жизнь?;
- Riekelt H. Houtkooper, Laurent Mouchiroud, Dongryeol Ryu, Norman Moullan, Elena Katsyuba, et. al.. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 497, 451-457;
- Эпигенетика: невидимый командир генома;
- Старение и долголетие: эпигеном раскрывает тайны;
- Stephen W. G. Tait, Douglas R. Green. (2012). Mitochondria and cell signalling. J Cell Sci. 125, 807-815;
- Patries M. Herst, Matthew R. Rowe, Georgia M. Carson, Michael V. Berridge. (2017). Functional Mitochondria in Health and Disease. Front. Endocrinol.. 8;
- A Oberst, C Bender, D R Green. (2008). Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 15, 1139-1146;
- Апоптоз, или Путь самурая;
- Толл-подобные рецепторы: от революционной идеи Чарльза Джейнуэя до Нобелевской премии 2011 года;
- Иммунологическая Нобелевская премия (2011);
- Christine Vazquez, Stacy M. Horner. (2015). MAVS Coordination of Antiviral Innate Immunity. J. Virol.. 89, 6974-6977;
- Zbigniew Wyżewski, Karolina P. Gregorczyk, Justyna Struzik, Marek Niemiałtowski, Lidia Szulc-Dąbrowska. (2016). MAVS protein and its interactions with hepatitis A, B and C viruses. Postepy Hig Med Dosw. 70, 14-24;
- Varnesh Tiku, Man-Wah Tan, Ivan Dikic. (2020). Mitochondrial Functions in Infection and Immunity. Trends in Cell Biology. 30, 263-275;
- Mariana J. Kaplan, Marko Radic. (2012). Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J.I.. 189, 2689-2695;
- Manali Mukherjee, Paige Lacy, Shigeharu Ueki. (2018). Eosinophil Extracellular Traps and Inflammatory Pathologies—Untangling the Web!. Front. Immunol.. 9;
- S. Yousefi, M. Morshed, P. Amini, D. Stojkov, D. Simon, et. al.. (2015). Basophils exhibit antibacterial activity through extracellular trap formation. Allergy. 70, 1184-1188;
- Lena Pernas, Camilla Bean, John C. Boothroyd, Luca Scorrano. (2018). Mitochondria Restrict Growth of the Intracellular Parasite Toxoplasma gondii by Limiting Its Uptake of Fatty Acids. Cell Metabolism. 27, 886-897.e4;
- T CHAVAKIS, A BIERHAUS, P NAWROTH. (2004). RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes and Infection. 6, 1219-1225;
- Qiuyun Liu, Danyan Zhang, Diyu Hu, Xiangmei Zhou, Yang Zhou. (2018). The role of mitochondria in NLRP3 inflammasome activation. Molecular Immunology. 103, 115-124;
- Bo-Zong Shao, Zhe-Qi Xu, Bin-Ze Han, Ding-Feng Su, Chong Liu. (2015). NLRP3 inflammasome and its inhibitors: a review. Front. Pharmacol.. 6;
- Rebecca C Coll, Avril A B Robertson, Jae Jin Chae, Sarah C Higgins, Raúl Muñoz-Planillo, et. al.. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 21, 248-255;
- Marta Gonzalez-Freire, Rafael de Cabo, Michel Bernier, Steven J. Sollott, Elisa Fabbri, et. al.. (2015). Reconsidering the Role of Mitochondria in Aging. GERONA. 70, 1334-1342;
- Dimitry A. Chistiakov, Igor A. Sobenin, Victor V. Revin, Alexander N. Orekhov, Yuri V. Bobryshev. (2014). Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Research International. 2014, 1-7;
- Nuo Sun, Richard J. Youle, Toren Finkel. (2016). The Mitochondrial Basis of Aging. Molecular Cell. 61, 654-666;
- Tobias Brandt, Arnaud Mourier, Luke S Tain, Linda Partridge, Nils-Göran Larsson, Werner Kühlbrandt. (2017). Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. eLife. 6;
- Timo E.S. Kauppila, Johanna H.K. Kauppila, Nils-Göran Larsson. (2017). Mammalian Mitochondria and Aging: An Update. Cell Metabolism. 25, 57-71;
- Bhupendra Singh, Trenton R. Schoeb, Prachi Bajpai, Andrzej Slominski, Keshav K. Singh. (2018). Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 9;
- Riikka H. Hämäläinen, Juan C. Landoni, Kati J. Ahlqvist, Steffi Goffart, Sanna Ryytty, et. al.. (2019). Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias. Nat Metab. 1, 958-965;
- Daniela Bakula, Morten Scheibye-Knudsen. (2020). MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol.. 8;
- Болезни и изменения клеточного метаболизма;
- Вослед Варбургу — последние достижения в изучении биоэнергетики рака;
- Диагностика патологий клеточного метаболизма при нейродегенеративных заболеваниях;
- Mateusz Dyla, Daniel S. Terry, Magnus Kjaergaard, Thomas L.-M. Sørensen, Jacob Lauwring Andersen, et. al.. (2017). Dynamics of P-type ATPase transport revealed by single-molecule FRET. Nature. 551, 346-351;
- Rodrigue ROSSIGNOL, Benjamin FAUSTIN, Christophe ROCHER, Monique MALGAT, Jean-Pierre MAZAT, Thierry LETELLIER. (2003). Mitochondrial threshold effects. Biochemical Journal. 370, 751-762;
- Sumit Parikh, Amy Goldstein, Mary Kay Koenig, Fernando Scaglia, Gregory M. Enns, et. al.. (2015). Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 17, 689-701;
- Kumar Sharma. (2017). Mitochondrial Dysfunction in the Diabetic Kidney. Advances in Experimental Medicine and Biology. 553-562;
- Dong Hee Kim, Jeong-An Gim, Dahye Yoon, Suhkmann Kim, Heui-Soo Kim. (2017). Metabolomics and mitochondrial dysfunction in Alzheimer’s disease. Genes Genom. 39, 295-300;
- N Yoshimi, T Futamura, S E Bergen, Y Iwayama, T Ishima, et. al.. (2016). Cerebrospinal fluid metabolomics identifies a key role of isocitrate dehydrogenase in bipolar disorder: evidence in support of mitochondrial dysfunction hypothesis. Mol Psychiatry. 21, 1504-1510;
- Wynn G. Hunter, Jacob P. Kelly, Robert W. McGarrah, Michel G. Khouri, Damian Craig, et. al.. (2016). Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure With Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. JAHA. 5;
- Hannah E Steele, Rita Horvath, Jon J Lyon, Patrick F Chinnery. (2017). Monitoring clinical progression with mitochondrial disease biomarkers. Brain. 140, 2530-2540;
- Tianhong Su, Doug Turnbull, Laura Greaves. (2018). Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes. 9, 182;
- S MOHAMED, T HANKE, A ERASMI, M BECHTEL, M SCHARFSCHWERDT, et. al.. (2006). Mitochondrial DNA deletions and the aging heart. Experimental Gerontology. 41, 508-517;
- Обо всех РНК на свете, больших и малых;
- Michelangelo Mancuso, Daniele Orsucci, Corrado Angelini, Enrico Bertini, Valerio Carelli, et. al.. (2014). The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender?. J Neurol. 261, 504-510;
- Bhupendra Singh, Trenton R. Schoeb, Prachi Bajpai, Andrzej Slominski, Keshav K. Singh. (2018). Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 9;
- O’Hara R., Tedone E., Ludlow A., Huang E., Arosio B., Mari D., Shay J.W. (2019). Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single cell resolution: a focus on aging and cancer. bioRxiv;
- Kirsty Foote, Johannes Reinhold, Emma P. K. Yu, Nichola L. Figg, Alison Finigan, et. al.. (2018). Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell. 17, e12773;
- Leila Zabihi Diba, Seyed Mojtaba Mohaddes Ardebili, Jalal Gharesouran, Massoud Houshmand. (2016). Age-related decrease in mtDNA content as a consequence of mtDNA 4977 bp deletion. Mitochondrial DNA Part A. 27, 3008-3012;
- Sophia Miliotis, Bryan Nicolalde, Mayra Ortega, Jackie Yepez, Andrés Caicedo. (2019). Forms of extracellular mitochondria and their impact in health. Mitochondrion. 48, 16-30;
- Changwan Ryu, Huanxing Sun, Mridu Gulati, Jose D. Herazo-Maya, Yonglin Chen, et. al.. (2017). Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 196, 1571-1581;
- C. Meißner, N. von Wurmb, M. Oehmichen. (1997). Detection of the age-dependent 4977 bp deletion of mitochondrial DNA. International Journal of Legal Medicine. 110, 288-291;
- L. A. Zinovkina, R. A. Zinovkin. (2015). DNA methylation, mitochondria, and programmed aging. Biochemistry Moscow. 80, 1571-1577;
- Vito Iacobazzi, Alessandra Castegna, Vittoria Infantino, Generoso Andria. (2013). Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Molecular Genetics and Metabolism. 110, 25-34;
- Ladislav Valkovič, Marek Chmelík, Martin Krššák. (2017). In-vivo 31 P-MRS of skeletal muscle and liver: A way for non-invasive assessment of their metabolism. Analytical Biochemistry. 529, 193-215;
- 12 методов в картинках: нейробиология;
- Joel S Riley, Stephen WG Tait. (2020). Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21;
- Lance H. Rodan, Greg D. Wells, Laura Banks, Sara Thompson, Jane E. Schneiderman, Ingrid Tein. (2015). L-Arginine Affects Aerobic Capacity and Muscle Metabolism in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-Like Episodes) Syndrome. PLoS ONE. 10, e0127066;
- J Finsterer, S Shorny, J Capek, C Cerny-Zacharias, B Pelzl, et. al.. (1998). Lactate stress test in the diagnosis of mitochondrial myopathy. Journal of the Neurological Sciences. 159, 176-180;
- Josef Finsterer, Erika Milvay. (2002). Lactate Stress Testing in 155 Patients with Mitochondriopathy. Can. j. neurol. sci.. 29, 49-53;
- Mark Tarnopolsky. (2004). Exercise testing as a diagnostic entity in mitochondrial myopathies. Mitochondrion. 4, 529-542;
- Sara Cogliati, Jose A. Enriquez, Luca Scorrano. (2016). Mitochondrial Cristae: Where Beauty Meets Functionality. Trends in Biochemical Sciences. 41, 261-273;
- Jesus R. Huertas, Rafael A. Casuso, Pablo Hernansanz Agustín, Sara Cogliati. (2019). Stay Fit, Stay Young: Mitochondria in Movement: The Role of Exercise in the New Mitochondrial Paradigm. Oxidative Medicine and Cellular Longevity. 2019, 1-18;
- Abdul Khalique, Rucha D. Sarwade, Poonam R. Pandey, M.V. Vijayakumar, Manoj K. Bhat, Vasudevan Seshadri. (2016). Prolonged exposure to insulin with insufficient glucose leads to impaired Glut4 translocation. Biochemical and Biophysical Research Communications. 474, 64-70;
- F. Tremblay, C. Lavigne, H. Jacques, A. Marette. (2001). Defective Insulin-Induced GLUT4 Translocation in Skeletal Muscle of High Fat-Fed Rats Is Associated With Alterations in Both Akt/Protein Kinase B and Atypical Protein Kinase C ( / ) Activities. Diabetes. 50, 1901-1910;
- H. Zong, J. M. Ren, L. H. Young, M. Pypaert, J. Mu, et. al.. (2002). AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proceedings of the National Academy of Sciences. 99, 15983-15987;
- Adam L. Bujak, Justin D. Crane, James S. Lally, Rebecca J. Ford, Sally J. Kang, et. al.. (2015). AMPK Activation of Muscle Autophagy Prevents Fasting-Induced Hypoglycemia and Myopathy during Aging. Cell Metabolism. 21, 883-890;
- Микробиом кишечника: мир внутри нас;
- Рожденные кесаревым сечением младенцы не получают ключевых микробов?;
- Микробные фармацевты внутри нас. Человеческий микробиом — спаситель и убийца;
- Jason Lloyd-Price, IBDMDB Investigators, Cesar Arze, Ashwin N. Ananthakrishnan, Melanie Schirmer, et. al.. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 569, 655-662;
- Alfredo Franco-Obregón, Jack A. Gilbert. (2017). The Microbiome-Mitochondrion Connection: Common Ancestries, Common Mechanisms, Common Goals. mSystems. 2;
- Sandra M. Cardoso, Nuno Empadinhas. (2018). The Microbiome-Mitochondria Dance in Prodromal Parkinson’s Disease. Front. Physiol.. 9;
- Dakota N. Jackson, Arianne L. Theiss. (2020). Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes. 11, 285-304;
- Bindu D. Paul, Solomon H. Snyder, Khosrow Kashfi. (2021). Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biology. 38, 101772;
- Dongryeol Ryu, Laurent Mouchiroud, Pénélope A Andreux, Elena Katsyuba, Norman Moullan, et. al.. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 22, 879-888;
- Xiao-Xiao Li, Bun Tsoi, Yi-Fang Li, Hiroshi Kurihara, Rong-Rong He. (2015). Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J Histochem Cytochem.. 63, 301-311;
- Maria J. Redlak, Jacinda J. Power, Thomas A. Miller. (2005). Role of mitochondria in aspirin-induced apoptosis in human gastric epithelial cells. American Journal of Physiology-Gastrointestinal and Liver Physiology. 289, G731-G738;
- Federico Pietrocola, Francesca Castoldi, Maria Markaki, Sylvie Lachkar, Guo Chen, et. al.. (2018). Aspirin Recapitulates Features of Caloric Restriction. Cell Reports. 22, 2395-2407;
- Radha Uppala, Brianne Dudiak, Megan E. Beck, Sivakama S. Bharathi, Yuxun Zhang, et. al.. (2017). Aspirin increases mitochondrial fatty acid oxidation. Biochemical and Biophysical Research Communications. 482, 346-351;
- Alexander A. Soukas, Haibin Hao, Lianfeng Wu. (2019). Metformin as Anti-Aging Therapy: Is It for Everyone?. Trends in Endocrinology & Metabolism. 30, 745-755;
- Leena P. Bharath, Madhur Agrawal, Grace McCambridge, Dequina A. Nicholas, Hatice Hasturk, et. al.. (2020). Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metabolism. 32, 44-55.e6;
- Adam R. Konopka, Jaime L. Laurin, Hayden M. Schoenberg, Justin J. Reid, William M. Castor, et. al.. (2019). Metformin inhibits mitochondrial adaptations to aerobic exercise training in older adults. Aging Cell. 18;
- Jesus R. Huertas, Rafael A. Casuso, Pablo Hernansanz Agustín, Sara Cogliati. (2019). Stay Fit, Stay Young: Mitochondria in Movement: The Role of Exercise in the New Mitochondrial Paradigm. Oxidative Medicine and Cellular Longevity. 2019, 1-18;
- Chengjie Song, Jun Zhang, Shasha Qi, Zhen Liu, Xiaoyang Zhang, et. al.. (2019). Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell. 18, e12941;
- Ya-Wen Lu, Steven M. Claypool. (2015). Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet.. 6;
- Wei-Wei Chen, Yu-Jen Chao, Wan-Hsin Chang, Jui-Fen Chan, Yuan-Hao Howard Hsu. (2018). Phosphatidylglycerol Incorporates into Cardiolipin to Improve Mitochondrial Activity and Inhibits Inflammation. Sci Rep. 8;
- Juan D. Chavez, Xiaoting Tang, Matthew D. Campbell, Gustavo Reyes, Philip A. Kramer, et. al.. (2020). Mitochondrial protein interaction landscape of SS-31. Proc Natl Acad Sci USA. 117, 15363-15373;
- Alexander V. Birk, Shaoyi Liu, Yi Soong, William Mills, Pradeep Singh, et. al.. (2013). The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. JASN. 24, 1250-1261;
- Sunghee Cho, Hazel H. Szeto, Eunhee Kim, Hyunjoo Kim, Aaron T. Tolhurst, John T. Pinto. (2007). A Novel Cell-permeable Antioxidant Peptide, SS31, Attenuates Ischemic Brain Injury by Down-regulating CD36. Journal of Biological Chemistry. 282, 4634-4642;
- Fan-Yen Lee, Pei-Lin Shao, Christopher Wallace, Sarah Chua, Pei-Hsun Sung, et. al.. (2018). Combined Therapy with SS31 and Mitochondria Mitigates Myocardial Ischemia-Reperfusion Injury in Rats. IJMS. 19, 2782;
- Michael P. Siegel, Shane E. Kruse, Justin M. Percival, Jorming Goh, Collin C. White, et. al.. (2013). Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 12, 763-771;
- Stefano Tarantini, Noa M. Valcarcel-Ares, Andriy Yabluchanskiy, Gabor A. Fulop, Peter Hertelendy, et. al.. (2018). Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 17, e12731;
- Matthew J. Rossman, Jessica R. Santos-Parker, Chelsea A.C. Steward, Nina Z. Bispham, Lauren M. Cuevas, et. al.. (2018). Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension. 71, 1056-1063;
- Tomoyasu Kadoguchi, Shintaro Kinugawa, Shingo Takada, Arata Fukushima, Takaaki Furihata, et. al.. (2015). Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol. 100, 312-322;
- Sara Conti, Paola Cassis, Ariela Benigni. (2012). Aging and the Renin-Angiotensin System. Hypertension. 60, 878-883;
- Rita Valenzuela, Maria A Costa-Besada, Javier Iglesias-Gonzalez, Emma Perez-Costas, Begoña Villar-Cheda, et. al.. (2016). Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 7, e2427-e2427;
- Генная терапия: познакомьтесь с лекарствами будущего;
- Трое в лодке: о легализации замены митохондрий;
- Carlos T Moraes. (2014). A magic bullet to specifically eliminate mutated mitochondrial genomes from patients' cells. EMBO Mol Med. 6, 434-435;
- Alexander N. Patananan, Ting-Hsiang Wu, Pei-Yu Chiou, Michael A. Teitell. (2016). Modifying the Mitochondrial Genome. Cell Metabolism. 23, 785-796;
- S R Bacman, S L Williams, S Garcia, C T Moraes. (2010). Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 17, 713-720;
- S R Bacman, S L Williams, D Duan, C T Moraes. (2012). Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 19, 1101-1106;
- Pradeep Reddy, Alejandro Ocampo, Keiichiro Suzuki, Jinping Luo, Sandra R. Bacman, et. al.. (2015). Selective Elimination of Mitochondrial Mutations in the Germline by Genome Editing. Cell. 161, 459-469;
- Jens Boch, Ulla Bonas. (2010). XanthomonasAvrBs3 Family-Type III Effectors: Discovery and Function. Annu. Rev. Phytopathol.. 48, 419-436;
- Masami Hashimoto, Sandra R Bacman, Susana Peralta, Marni J Falk, Anne Chomyn, et. al.. (2015). MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Molecular Therapy. 23, 1592-1599;
- Sandra R. Bacman, Johanna H. K. Kauppila, Claudia V. Pereira, Nadee Nissanka, Maria Miranda, et. al.. (2018). MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med. 24, 1696-1700;
- Просто о сложном: CRISPR/Cas;
- РНК, ножницы, геномы: объявлены лауреаты Нобелевской премии по химии 2020;
- Северинов К. (2016). Редактирование генома с CRISPR/Cas9. «Постнаука»;
- Chu-Yuan Chang, Min-Zong Liang, Linyi Chen. (2019). Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl Neurodegener. 8;
- Daniel Torralba, Francesc Baixauli, Francisco Sánchez-Madrid. (2016). Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol.. 4;
- Kazuhide Hayakawa, Elga Esposito, Xiaohua Wang, Yasukazu Terasaki, Yi Liu, et. al.. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 535, 551-555;
- Adrien Acquistapace, Thierry Bru, Pierre-François Lesault, Florence Figeac, Amélie E. Coudert, et. al.. (2011). Human Mesenchymal Stem Cells Reprogram Adult Cardiomyocytes Toward a Progenitor-Like State Through Partial Cell Fusion and Mitochondria Transfer. STEM CELLS. 29, 812-824;
- Yi-Chao Hsu, Yu-Ting Wu, Ting-Hsien Yu, Yau-Huei Wei. (2016). Mitochondria in mesenchymal stem cell biology and cell therapy: From cellular differentiation to mitochondrial transfer. Seminars in Cell & Developmental Biology. 52, 119-131;
- Megan V. Jackson, Thomas J. Morrison, Declan F. Doherty, Daniel F. McAuley, Michael A. Matthay, et. al.. (2016). Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells. 34, 2210-2223;
- Andrés Caicedo, Pedro M. Aponte, Francisco Cabrera, Carmen Hidalgo, Maroun Khoury. (2017). Artificial Mitochondria Transfer: Current Challenges, Advances, and Future Applications. Stem Cells International. 2017, 1-23;
- J. A. Barritt, C. A. Brenner, H. E. Malter, J. Cohen. (2001). Mitochondria in human offspring derived from ooplasmic transplantation: Brief communication. Human Reproduction. 16, 513-516;
- Трое в лодке: о легализации замены митохондрий;
- J Cohen. (1998). Ooplasmic transfer in mature human oocytes. Molecular Human Reproduction. 4, 269-280;
- Andrés Caicedo, Pedro M. Aponte, Francisco Cabrera, Carmen Hidalgo, Maroun Khoury. (2017). Artificial Mitochondria Transfer: Current Challenges, Advances, and Future Applications. Stem Cells International. 2017, 1-23;
- Chu-Yuan Chang, Min-Zong Liang, Linyi Chen. (2019). Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl Neurodegener. 8;
- James D. McCully, Sidney Levitsky, Pedro J. Nido, Douglas B. Cowan. (2016). Mitochondrial transplantation for therapeutic use. Clinical and Translational Medicine. 5;
- James D. McCully, Douglas B. Cowan, Sitaram M. Emani, Pedro J. del Nido. (2017). Mitochondrial transplantation: From animal models to clinical use in humans. Mitochondrion. 34, 127-134;
- Akihiro Masuzawa, Kendra M. Black, Christina A. Pacak, Maria Ericsson, Reanne J. Barnett, et. al.. (2013). Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. American Journal of Physiology-Heart and Circulatory Physiology. 304, H966-H982.



Комментарии
0Чтобы оставить комментарий, необходимо
войти