Одноклеточное секвенирование: разделяй, изучай и властвуй
13 августа 2021
Одноклеточное секвенирование: разделяй, изучай и властвуй
- 5990
- 0
- 17
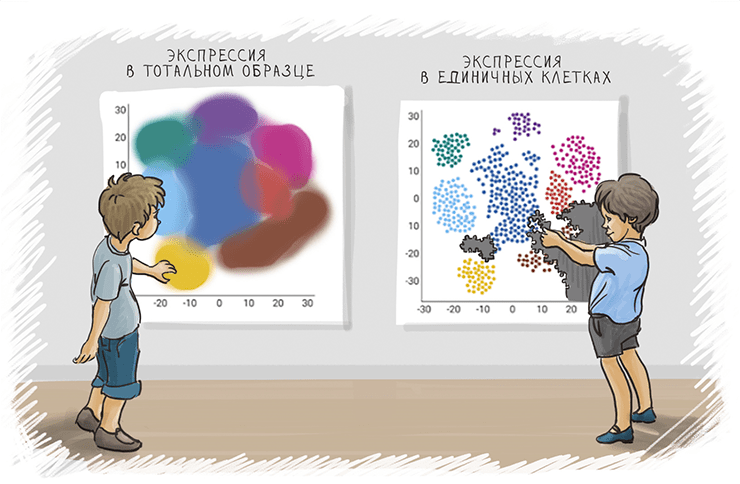
Одноклеточное секвенирование дает возможность увидеть более детальную картинку. Рисунок в полном размере.
иллюстрация Елены Беловой
-
Автор
-
Редакторы
-
Иллюстратор
Темы
Современные методы полногеномного секвенирования позволяют считывать миллиарды последовательностей нуклеиновых кислот (НК) — ДНК или РНК — за считанные часы. Человечество вплотную подобралось к пониманию механизмов реализации генетического материала в живых системах, и новые технологии одноклеточного секвенирования с возможностью визуализации пространственного положения нуклеиновых кислот внутри или вне клетки, похоже, стали еще одним шагом к этой цели.

Ультрасовременные методы
Методы, которыми оперирует современная наука, постоянно совершенствуются. Некоторые области развиваются настолько стремительно, что порой даже специалистам сложно уследить за новейшими приборами и модификациями методик. Так происходит, например, в геномике. Другие же области (такие как гистология и связанные с ней световая и флуоресцентная микроскопия) развиваются куда медленнее, но всё равно расширяются и обзаводятся более высокопроизводительными методами. В статьях нового спецпроекта (он продолжает наш хит — «12 биологических методов в картинках») мы хотим рассказать, какие методы помогают ученым совершать новые открытия сегодня, и чего нам ждать в будущем.
Партнер этой статьи — «СкайДжин».
Пять причин выбрать SkyGen:
- Доступ к высококачественной продукции для молекулярной биологии.
- Высококвалифицированная научная поддержка.
- Быстрая логистика и складская программа.
- Удобное и взаимовыгодное сотрудничество.
- Адекватные цены.
К середине прошлого столетия, в момент открытия нуклеиновых кислот (НК — ДНК и РНК) как носителей генетической информации живых организмов, перед исследователями встал вопрос о возможности прочтения их последовательности — секвенирования. Десятки лет и поступательный прогресс во многих отраслях науки и инженерии сделали возможным сначала определение последовательности отдельных фрагментов ДНК, а затем и секвенирование целых геномов.
К настоящему времени разработано и выведено на биотехнологический рынок значительное количество методов секвенирования НК. Самый популярный и надежный из них — секвенирование методом «терминаторов» — разработал британский биолог Фредерик Сэнгер с коллегами в далеком 1977 году [1]. За десятки лет данный метод неоднократно модифицировался (например, радиоактивные метки заменили флуоресцентными), однако идея, лежащая в его основе, не изменилась. Анализируемый, предварительно многократно амплифицированный фрагмент ДНК (до 1000 пар нуклеотидов (п.н.) — букв генетического алфавита) используется как матрица для синтеза комплементарной (дочерней) цепочки ферментом ДНК-полимеразой в присутствии дезоксинуклеозидтрифосфатов (А, Т, Г, Ц — аденина, тимина, гуанина и цитозина). Кроме того, в смеси находятся флуоресцентно меченные дидезоксинуклеозидтрифосфаты, которые прерывают реакцию синтеза из-за особенности химического строения. Благодаря флуоресценции и электрофорезу в геле можно точно установить длину конкретного секвенируемого фрагмента ДНК и последнюю букву в нем. Поскольку в реакционной смеси параллельно проходят тысячи реакций, используя этот уже полностью автоматизированный метод, можно восстановить последовательности десятков ДНК-фрагментов.
Несмотря на то, что определение последовательностей НК по Сэнгеру до сих пор является «золотым» стандартом секвенирования и нашло свое применение в различных жизненно важных отраслях (таких как медицинская генетика и криминалистика), его производительность оставляет желать лучшего [2], [3].
Геномы многих растений и позвоночных животных, в том числе и человека, состоят из миллиардов «букв», и определение их полных последовательностей с использованием настолько «олдскульных» методов экономически нецелесообразно.
Не зря в начале XXI века в дополнение к сэнгеровскому секвенированию появилась целая плеяда методов, позволяющих считывать миллионы и миллиарды коротких ДНК-фрагментов (длиной от 25 до 500 п.н.) одновременно: они получили название секвенирования нового поколения (next-generation sequencing, NGS). В отличие от секвенирования по Сэнгеру, методы NGS позволяют производить глубокое (многократное) прочтение генетического материала, которое необходимо, например, для ресеквенирования геномов, транскриптомных (анализ экспрессии белок-кодирующих генов и некодирующих РНК) и эпигеномных (метилирование и гидроксиметилирование ДНК, метилирование РНК) исследований . Такой рост производительности привел к возможности определения последовательности сразу десятков геномов (в зависимости от их размера), транскриптомов или эпигеномов за один запуск прибора [2].
Подробнее с тем, какие возможности предоставляют ученым современные подходы к расшифровке генома, читатель может ознакомиться в статьях: «454-секвенирование (высокопроизводительное пиросеквенирование ДНК» [4], «Код жизни: прочесть не значит понять» [5], «Новый взгляд на геном: не просто цепочка генов, а трехмерная сеть, интегрирующая функциональные домены ядра» [6], «Неуловимая архитектура хроматина мухи» [7], «Молекулы и эпигеном» [8], «Загадочное путешествие некодирующей РНК Xist по Х-хромосоме» [9].
За последние годы сразу несколько технологий глубокого секвенирования внедрились в современную биологическую науку. Некоторые из них — лигазное секвенирование или пиросеквенирование, о которых можно почитать в нашей обзорной статье «12 методов в картинках: секвенирование нуклеиновых кислот» [2], — уже не поддерживаются их разработчиками, в то время как секвенирование на молекулярных кластерах (также известное как технология Illumina), или одномолекулярное секвенирование в реальном времени (чуть позднее к ним присоединилась технология cPAS секвенирования), прочно заняли свои ниши. Одна часть этих технологий прекрасно подходит для анализа новых, ранее не опубликованных геномов или поиска длинных некодирующих РНК, тогда как другая эффективна и обходится дешевле при ресеквенировании или анализе экспрессии генов. Более того, исследователи могут комбинировать разнообразные методы для достижения наиболее интересных результатов. В частности, одномолекулярное секвенирование — которое осуществляется благодаря приборам PacBio, — также как и нанопоровое секвенирование (Oxford Nanopore) [2], [10], регулярно используется вместе с технологией Illumina для хромосомной сборки новых геномов, в том числе и полиплоидных [11–13].
Одноклеточное секвенирование (ОКС, scRNA-seq)
Таким образом, на сегодняшний день секвенирование ДНК и РНК — это во многом рутинная процедура, которая служит лишь в качестве одного из множества методов современной молекулярной биологии и биотехнологии. Используемые сегодня методы секвенирования НК позволяют оценивать не только последовательность ДНК, но и экспрессию кодируемых ею молекул РНК (транскриптом), а также разнообразные типы модификаций генома и транскриптома, такие как метилирование, тесно связанное с активностью генов. В то же время перед учеными то и дело встают новые задачи. Так, несмотря на генетическую идентичность клеток в одном организме, иногда требуется оценить разницу между одиночными клетками и их субпопуляциями; более того, последние исследования показали, что из ста триллионов клеток, составляющих человеческое тело, не найдется и двух с полностью одинаковым геномом — из-за мутаций, накапливающихся в эмбриогенезе и в течение жизни организма [14].
Дело в том, что большинство широко применяемых методов анализа нуклеиновых кислот позволяет работать с тканями, которые состоят из абсолютно разных клеточных субтипов. При этом, анализируя образцы ДНК или РНК из таких экстрактов, ученые получают некие усредненные результаты и редко могут прицельно дотянуться до конкретной функции или механизма.
В результате исследователи, обращаясь к классическим методам глубокого секвенирования, нередко сталкиваются с трудностями — например, при анализе геномов некультивируемых микроорганизмов, изучении генетического мозаицизма [14] или геномном анализе клеточных субпопуляций раковых опухолей. Связаны эти трудности, как правило, с биоинформатическим анализом данных, а иногда эксперименты вовсе не дают необходимых результатов [15]. Не меньше проблем у ученых до недавнего времени возникало в ходе анализа экспрессии генов или метилирования в комплексных тканях, состоящих из разных типов клеток (например, тканях головного мозга), или в тканях, пронизанных кровеносными сосудами [16], [17].
Ответом на все эти вызовы стали технологии одноклеточного секвенирования (ОКС). Они позволяют получать информацию о многомерности тканей, выявляя различия в клеточных популяциях, и оценивать клеточные взаимосвязи, а не получать лишь среднее значение для многих клеток, теряя информацию об их гетерогенности, как это было раньше. И если еще недавно широкое распространение ОКС сдерживалось его высокой стоимостью, то по мере появления новых методов пробоподготовки и секвенирования сегодня данный подход используется всё чаще [18].
Одноклеточное секвенирование можно разделить на четыре важнейших этапа:
- Подготовка анализируемого биологического образца, изоляция и «обогащение» единичных клеток. Приготовление суспензии клеток.
- Выделение ДНК или РНК. Приготовление ДНК/РНК-библиотек для секвенирования.
- Секвенирование полученных библиотек с использованием выбранной пользователем платформы.
- Биоинформатический анализ данных и их визуализация.
На протяжении всех четырех этапов важно обращать внимание на ряд экспериментальных параметров и критических точек, следование которым позволит провести успешный эксперимент и получить достоверные данные [19].
Пожалуй, одним из важнейших этапов при одноклеточном секвенировании является пробоподготовка, поскольку она может в значительной мере повлиять на результат. Например, ткани с высокими показателями адгезии между клетками должны быть тщательно диссоциированы до одноклеточного уровня. С другой стороны, экспериментатор должен отдавать себе отчет в том, что более разрушительные методы суспендирования клеток могут иметь фатальные последствия для результатов, приводя к гибели части клеток и тем самым нарушая соотношения субпопуляций в ткани. На сегодняшний день для диссоциации тканей используют три основных подхода: ферментативный (обработка биологического образца ферментами, разрушающими межклеточный матрикс, — коллагеназой, эластазой, трипсином и другими), механический (диссекция клеток, пипетирование, центрифугирование и др.), а также комбинированный, который включает в себя первые два.
Кроме того, в некоторых случаях необходима не только диссоциация клеток и приготовление их суспензий, но и «обогащение» редко представленных клеточных субпопуляций в общем клеточном пуле, а также элиминация мертвых клеток. Для этих случаев может быть использована целая плеяда методов, которые позволяют идентифицировать определенную субпопуляцию клеток, основываясь на их массе, размере, плотности. Различные флуоресцентные метки, в свою очередь, применяются для идентификации и последующей сортировки определенных типов клеток [20].
Широкое распространение при подготовке биологических образцов к ОКС получили методы проточной цитофлуориметрии. Проточная цитометрия является ценным инструментом для контроля качества, поскольку с ее помощью одновременно можно оценивать множество показателей, включая размер клеток, их жизнеспособность, а также адгезию между клетками. Подробнее о методах цитометрии можно почитать в ранее опубликованной на «Биомолекуле» статье «12 методов в картинках: проточная цитофлуориметрия» [21].
Последние достижения в области микрофлюидики позволяют осуществлять высокопроизводительный генетический анализ на уровне отдельных клеток. Технологии, представленные в настоящее время на рынке, дают возможность эффективно анализировать генетические особенности десятков тысяч клеток в течение одного эксперимента. Высокой популярностью пользуются методы, при помощи которых клетки сепарируются в проточных каналах на специальных чипах (технология Fluidigm, детально описанная в заметке: «Опухолевые разговоры, или Роль микроокружения в развитии рака» [22]), а также технология разделения отдельных клеток путем их инкапсулирования в виде индивидуальных эмульсионных капель с гелевыми частицами, содержащими уникальный баркод, позволяющий мультиплексировать изучаемую популяцию клеток (технология 10x Genomics, на основе которой вышло уже более 2200 рецензируемых статей, 200 из которых опубликованы таких топовых журналах, как: Cell, Nature, Science). Таким образом, каждая клетка получает свой первичный идентификатор, который далее преобразуется в (эпи)генетический паспорт [23].
После лизиса из каждой клетки выделяется генетический материал, используемый для приготовления ДНК-библиотек. Поскольку каждая клетка содержит лишь один диплоидный геном, его необходимо предварительно амплифицировать, так как современные платформы для секвенирования не позволяют работать с одной или двумя копиями генома (рис. 2). Для наработки достаточного количества копий при пробоподготовке используют разнообразные методы полногеномной амплификации (whole genome amplification, WGA), позволяющие избежать значительного количества ошибок, — например, изотермическую ПЦР. В случае работы с различными типами РНК пул транскриптов предварительно переводят в комплементарную ДНК (кДНК) при помощи обратной транскриптазы, а затем также амплифицируют отдельно для каждой клетки. В настоящее время особой популярностью по-прежнему пользуется метод амплификации со множественным замещением цепи (multiple displacement amplification, MDA) с применением полимеразы phi29 (выделенной из одноименного бактериофага). Данная полимераза славится своей высокой процессивностью, малым количеством ошибок и широтой покрытия генома (около 84%) [24].
Другой метод полногеномной амплификации, известный под аббревиатурой MALBAC (Multiple annealing and looping-based amplification cycles), объединяет в себе MDA и обычную ПЦР и дает возможность не только амплифицировать значительную часть генома (более 50%) или транскриптома, но и добиться высокой равномерности покрытия и меньшей частоты потери аллелей относительно использования полимеразы phi29 [24] — она предпочтительно амплифицирует один аллель в гетерозиготном состоянии.
Таким образом, MDA и MALBAC имеют как преимущества, так и недостатки, а поэтому должны применяться в зависимости от целей эксперимента.

Рисунок 2. Основные этапы подготовки образцов ДНК и РНК для одноклеточного секвенирования
иллюстрация Елены Беловой по [51]
После выделения и амплификации НК для каждой анализируемой клетки исследователь должен выбрать дальнейшее направление пробоподготовки, которая в будущем определит методику секвенирования. Сегодня на биотехнологическом рынке представлены самые разные технологии определения последовательности НК, способные решать стоящие перед исследователем задачи. Вот главные из них:
- технология ионного полупроводникового секвенирования (Thermo Fisher Scientific);
- технология секвенирования на молекулярных кластерах с использованием флуоресцентно меченых нуклеотидов (Illumina);
- одномолекулярное секвенирование SMRT (Pacific Biosciences);
- нанопоровое секвенирование (Oxford Nanopore Technologies) [10];
- технология cPAS-секвенирования (BGI Genomics).
Первые четыре технологии подробно рассмотрены ранее на «Биомолекуле» («12 методов в картинках: секвенирование нуклеиновых кислот» [2] и «Нанопоровое секвенирование: на пороге третьей геномной революции» [10]), тогда как технология сPAS, выведенная на рынок коммерческих приборов китайской компанией BGI Genomics, вошла в повседневный обиход лишь несколько лет назад, но уже слегка потеснила технологию массового секвенирования, предлагаемую компанией Illumina.
На примере работы с геномной ДНК суть метода cPAS (рис. 3) заключается в следующем. К обоим концам предварительно фрагментированной ДНК (около 500 пар оснований) лигируют адаптеры. Затем полученную библиотеку циркуляризируют и амплифицируют, используя ранее упомянутый метод с множественным замещением цепи (MDA) по принципу катящегося кольца, то есть исходный фрагмент прочитывается многократно. Такой подход позволяет избежать клонального накопления ошибок полимеразы. После того, как количество амплифицированных фрагментов достигнет нескольких сотен, они укладываются с использованием запатентованной технологии в ДНК-наношарик (DNA nanoball, DNB) [25].

Рисунок 3. Технология cPAS-секвенирования. Фрагментированная геномная ДНК лигируется с четырьмя адаптерами, формируя кольцевую ДНК-библиотеку. ДНК-наношарик формируется путем репликации по принципу катящегося кольца. Каждый ДНК-наношарик помещается в определенное место на ячейке секвенатора. Считывание ДНК осуществляется с использованием принципов гибридизации и лигазного секвенирования.
[52], рисунок адаптирован
Полученные ДНК-фрагменты, компактизированные в виде ДНК-наношариков, наносятся на проточную ячейку секвенатора, где закрепляются за счет электростатических сил. На проточной ячейке проводят циклический процесс секвенирования, в котором объединяются принципы гибридизации и лигазного секвенирования. Реакционная смесь для синтеза комплементарной ДНК подается на поверхность проточной ячейки и содержит ферменты, а также четыре типа флуоресцентно меченых проб, состоящих из нуклеотидов универсальной специфичности (способных связаться с любым из нуклеотидов матричной ДНК), а также одного известного нуклеотида (A, T, G или С) на 5′-конце. После гибридизации пробы и специфичного нуклеотида лигирования по 5′-концу камерой с ПЗС-матрицей считывается флуоресценция пробы, а затем универсальные нуклеотиды и флуоресцентный краситель смываются. Эта серия шагов продолжается определенное количество раз, число которых задает пользователь [25].
Биоинформатический анализ данных одноклеточного секвенирования
Биоинформатический анализ данных одноклеточного секвенирования, несмотря на идеологическое сходство с классическим, рутинным анализом транскриптома в тканях и клеточных культурах (подробная информация представлена в классическом обзоре: A survey of best practices for RNA-seq data analysis [26]), представляет собой не самую тривиальную часть эксперимента, и в рамках данного обзора крайне сложно пройтись по всем подводным камням, которые ожидают компьютерного биолога. Уж слишком много приложений одноклеточного секвенирования существует сейчас, да и каждое конкретное исследование, ткань или модель требуют особого подхода.
Важный момент при анализе данных одноклеточного секвенирования — необходимость нормализации изучаемых образцов. Дело в том, что в ходе экспериментальных процедур (сепарация клеток, выделение НК, полногеномная амплификация и создание библиотек) существует значительная вероятность ошибок. Эти, казалось бы, незначительные этапы в дальнейшем могут существенно влиять на ширину и глубину покрытия генома/транскриптома, на смещение в числе определенных типов клеток и их субпопуляций, а также на соотношение аллелей. Данные проблемы решаются воспроизводимостью экспериментальных процедур, добавлением технических и биологических повторностей, а также использованием дополнительных математических методов [19].
При геномном секвенировании глубина покрытия отдельных клеток значительно меньше, чем в массовых экспериментах. Таким образом, увеличение разрешения до клеточного уровня с использованием технологий ОКС означает также снижение достоверности обнаруживаемых механизмов, которые становятся более стабильными, когда суммируются (например, при анализе ткани). В конечном счете, задачи, которые ранее казались рутинными (например, поиск единичных нуклеотидных вариаций или геномных перестроек в субпопуляциях опухоли), требуют значительной методологической осторожности при обращении с данными одноклеточного секвенирования [27].
Так же как и при геномном секвенировании, анализ транскриптомных данных ОКС теоритически позволяет получить полное представление о взаимодействии транскриптов внутри отдельных клеток. Однако анализ экспрессии генов единичных клеток зачастую страдает от недостаточного покрытия, что в результате объединяет два различных типа нулевых значений в экспрессии конкретного транскрипта:
- Методологический шум, когда ген экспрессируется, но не обнаруживается с помощью технологии секвенирования.
- Отсутствие экспрессии данного транскрипта в конкретной клетке.
Во многом результаты транскриптомных исследований на одноклеточном уровне (впрочем, как и геномных) напрямую зависят от лабораторной части эксперимента, используемой платформы для пробоподготовки и секвенирования, глубины секвенирования, уровня экспрессии конкретного гена и так далее [27].
Подводя итог, можно сказать, что одноклеточное секвенирование НК может выявить сложные и редкие популяции клеток, найти регуляторные взаимосвязи между генами и отследить траектории развития отдельных клеточных линий. Дальнейшие технические усовершенствования на уровне молекулярной и клеточной биологии и доступных биоинформатических инструментов значительно облегчат как фундаментальные исследования, так и прикладное применение (медицина и биотехнология) этих технологий секвенирования [19].
Пространственная транскриптомика: новый шаг вглубь клетки и её машинерии
Распознавание пространственно-временных паттернов экспрессии генов имеет решающее значение для понимания основных биологических принципов от эмбриогенеза до возникновения и протекания болезней. Новые методы пространственной транскриптомики в единичных клетках расширяют наши знания о молекулярных процессах, протекающих внутри клеток и во внеклеточной среде. Эти подходы позволяют, например, реконструировать в цифровом виде паттерны эмбриональной экспрессии в 3D и успешно идентифицировать новые домены экспрессии, типы клеток и особенности тканей. Такие технологии открывают путь к беспристрастному и исчерпывающему анализу уровней экспрессии генов в пространственном и количественном выражении, способствуют пониманию функционирования генома и эпигенома в клетке.
В биологии пространственная концепция имеет большое значение, поскольку с ней можно описывать интерактивные биологические сети, в которых на каждый элемент влияет окружающая среда. Например, понимание молекулярных свойств отдельных клеток внутри многоклеточных организмов можно полностью понять только после того, как мы узнаем их физическое местонахождение. Причина в том, что клетки в различных тканевых субпопуляциях экспрессируют определенные наборы генов, поскольку они находятся под влиянием окружающих их клеток. Этот феномен управляет, например, формированием градиентов экспрессии генов вдоль основных осей эмбриона на разных стадиях развития. Эти градиенты затем направляют активацию генов, участвующих в эмбриональном развитии и необходимых для формирования органов. Другой пример — микроокружение опухоли, где несколько субпопуляций раковых клеток, составляющих опухоль, могут полностью отличаться друг от друга как по структурным особенностям, так и по профилю экспрессии генов . Осознание важности пространственной организации и точного положения молекулярных особенностей, исторически недостижимых в массовых и одноклеточных экспериментах, привело к разработке и внедрению методов пространственной транскриптомики [29], [30].
Подробнее о роли микроокружения в прогрессировании онкологических заболеваний можно почитать здесь: «Опухолевые разговоры, или Роль микроокружения в развитии рака» [22].
Современные методы молекулярной биологии, флуоресцентной гибридизации in situ (FISH) и микроскопии позволили не только анализировать синтез молекул РНК из отдельных тканей или отдельных клеток, но и визуализировать их в живых системах. Подобная визуализация in vivo может быть осуществлена посредством гибридизации меченого зонда, комплементарного транскриптам-мишеням. Более того, современные системы считывания анализа флуоресценции (например, сложные зонды, использующие комбинации флуорофоров) позволяют избегать спектрального перекрытия различных меток и тем самым анализировать значительное количество мишеней в клетке. В то же время этот подход не подходит для идентификации положения всех транскриптов в клетке [31].
Общей проблемой методов, основанных на FISH, считается их высокая цена и связанная с ней ограниченность в выборе транскриптов, а также высокий уровень фона аутофлуоресценции в непрозрачных образцах тканей [30].
Методы флуоресцентной гибридизации in situ, как показала практика, имеют существенные ограничения по числу реализующихся транскриптов, которые возможно описать. Поэтому были предложены другие методы, основанные на идентификации положения транскриптов в клетке, их захвата и последующего секвенирования ex situ. Такая концепция реализована в нескольких методологических подходах, где возможен анализ всего транскриптома. Однако основным препятствием для продвижения этих методов долгое время являлась ограниченная эффективность захвата РНК, которая становится всё более сложной при более высоком разрешении.
Первый способ пространственной транскриптомики был опубликован в 2016 году и в настоящее время коммерциализирован компанией 10x Genomics под названием 10x Visium. Суть данной методики заключается в приготовлении тонких (10–50 мкм) гистологических срезов и нанесении их на специальные предметные стекла с иммобилизованными на них поли-dT-олигонуклеотидами, для каждого из которых известно местоположение по Х- и Y-осям, и каждый из которых имеет специфический баркод. В дальнейшем ткань фиксируется, окрашивается и визуализируется (рис. 4).

Рисунок 4. Пример использования пространственной транскриптомики при анализе экспрессии генов в тканях префронтальной коры головного мозга человека. С помощью набора реагентов для пермеабилизации происходит снижение проницаемости клеточных мембран, и информационные РНК (матричные РНК, мРНК) диффундируют вниз к твердой поверхности слайда, локально гибридизуясь с иммобилизованными на его поверхности поли-dT-олигонуклеотидами. Затем методом обратной транскрипции проводится синтез кДНК и другие этапы подготовки библиотеки для последующего секвенирования с использованием одной из популярных платформ. Данные в дальнейшем экстраполируются на изображение среза ткани, полученное в начале эксперимента, предоставляя тем самым информацию о расположении конкретного транскрипта в клетке [32].
[53], рисунок адаптирован
Кроме описанного выше, существуют и другие методы пространственного секвенирования. Один из них (Slide-Seq) использует магнитные шарики с иммобилизованными поли-dT-олигонуклеотидами, на которых проходит считывание. Шарики случайно наносятся на слайд, и их местоположение декодируется перед началом нанесения на этот слайд образца [33]. Не менее интересной выглядит технология GeoMx, которая уже показала свою эффективность при работе со сложными образцами из парафиновых блоков. Кроме анализа экспрессии генов платформа GeoMx Digital Spatial Profiling (DSP) также предоставляет возможность пространственного профилирования белков. В процессе работы пользователь вручную выбирает интересующие его области клетки с помощью микроскопии. Затем эти области обрабатываются УФ-светом, вызывая высвобождение либо РНК-зонда связанного с транскриптом-мишенью, либо белка, помеченного антителом [34].
Биоинформатический анализ данных при пространственном транскриптомном анализе
Клетки разных типов пространственно и структурно организованы в тканях и выполняют свои разнообразные функции. Расшифровка сложной пространственной архитектуры гетерогенной ткани важна для понимания клеточных механизмов и функций в норме и при различных заболеваниях. Быстрое развитие технологий scRNA-seq позволило начать крупномасштабные исследования гетерогенных клеток и работ по отслеживанию родственных связей внутри ткани. К сожалению, из-за отсутствия пространственной информации scRNA-seq не способен идентифицировать структурную организацию гетерогенных клеток в сложной ткани. Поэтому в качестве дополнения к scRNA-seq были введены методы пространственного профилирования транскриптома. Чтобы выявить пространственную цитоархитектуру в тканях, используют описанные выше технологии, позволяющие проводить количественный анализ транскриптома с пространственным разрешением в отдельных срезах ткани. Технологические новшества и новые подходы в пространственной транскриптомике открыли неизведанный ландшафт, где транскрипционная информация помещается в пространственный контекст. Кластеризация — центральный компонент при анализе данных этого типа. Однако выбор количества кластеров для использования и интерпретация их взаимосвязей могут быть затруднительны [35], [36].
Новые технологии пространственной транскриптомики могут пространственно индексировать транскрипты и измерять профили экспрессии, улучшая наше понимание точной архитектуры тканей. Однако разрешение данных пространственной транскриптомики намного ниже, чем уровень одиночной клетки. Транскрипты, захваченные в определенном месте, обычно представляют смесь из 10–20 клеток. В результате измеренные экспрессии генов отражают хоть и небольшую, но смесь клеток. Следовательно, выявление клеточных составов в каждой точке данных ST имеет решающее значение для исследования молекулярной и клеточной архитектуры ткани с высоким разрешением [36].
К настоящему времени для кластеризации и визуализации данных разработано не так много программных продуктов, хотя определенные успехи в этой области уже появляются, в частности, с использованием искусственного интеллекта и математических моделей удалось определить расположение клеток и транскриптов, синтезируемых в них (рис. 6) [36].

Рисунок 6. Различные уровни разрешения и различные результаты, получаемые в зависимости от использованных методов
[19], рисунок адаптирован
Заключение
С появлением NGS-технологий в начале века человечество получило ключи и новые возможности для понимания молекулярных процессов, управляющих жизнью, однако анализ геномов, транскриптомов или эпигеномов представлялся серьезной проблемой при анализе комплексных тканей (кровь, нервная ткань) и органов, состоящих из различных субпопуляций клеток, различающихся по своему функционалу, а значит, и по тем генам, которые в них задействованы. Новые технологии одноклеточного секвенирования и пространственной транскриптомики придают новый импульс развитию современной геномики [18].
Технологии одноклеточного секвенирования дают уникальную возможность изучать молекулярную активность отдельных нервных клеток, визуализируя нейроны во времени и пространстве. Это позволяет изучать эволюцию головного мозга [37], идентифицировать различные типы нейронов и связанных с ними клеток в мозге. На сегодняшний день в головном мозге обнаружено большое число молекулярных типов нейронов, подтипов глиальных клеток [38], [39]. Ожидается, что понимание их работы на молекулярном уровне приблизит врачей к пониманию причины возникновения ряда заболеваний головного мозга, таких как шизофрения [40], болезни Альцгеймера [41] и Паркинсона [42].
Иммунная система играет важную роль в сопротивлении вторжению внешних патогенов. Многие ее клетки обладают уникальными функциями, и современная иммунология нуждается в технологиях одноклеточного секвенирования для более детального понимания молекулярных процессов, которые в них происходят . Секвенирование отдельных клеток иммунной системы поможет не только понять сложный иммунный ответ организма на различные патогены [43], но и оценить возможные новые пути борьбы с различными аллергическими реакциями у человека [44].
Подробнее о том, как современные ученые исследуют функции Т-лимфоцитов, читайте в статьях «Анализ индивидуальных репертуаров Т-клеточных рецепторов» [45], «Т-лимфоциты: путешественники и домоседы» [46] и «Тимоцитов ведут в зоопарк. Что такое эктопическая экспрессия и как она защищает нас от аутоиммунитета» [47].
Немаловажно использование технологий одноклеточного секвенирования в анализе причин и механизмов течения ряда генетически обусловленных заболеваний человека, в частности, онкологических. Исследования последних лет показали, что генетические или геномные вариации могут приводить к образованию клеток с различными характеристиками в опухолевой ткани, что делает опухолевую ткань очень гетерогенной. Такая высокая степень гетерогенности может быть связана с механизмами онкогенеза и метастазирования, поэтому исследования клеточных субпопуляций, населяющих опухоль, крайне важны [48]. Традиционные методы секвенирования позволяют получать среднее значение сигналов в опухолевой ткани, и генетическая неоднородность опухолевых клеток часто маскируется генетическим материалом наиболее представленной субпопуляции клеток опухоли. Технологии одноклеточного секвенирования могут прекрасно компенсировать недостатки традиционных методов расшифровки генома. Клеточная карта опухолевых клеток может быть построена путем обнаружения гетерогенности опухолевых клеток и пространственной транскриптомики. Технологии ОКС уже сейчас используются при исследовании различных опухолей и имеют большое значение для разработки новых методов диагностики и терапии [48], [49].
Анализ геномного разнообразия прокариот для комплексных образцов — также одна из основных мишеней одноклеточного секвенирования из-за сложности культивирования большинства микроорганизмов. Геномика единичных клеток — это мощный способ получения последовательностей геномов прокариот в отрыве от разработки сложных способов культивирования в лаборатории. Этот подход широко применяется к морским, почвенным, подземным, организменным и другим типам микробиомов для решения широкого круга вопросов, связанных с экологией и эволюцией микроорганизмов, здоровьем человека и потенциальным применением в биотехнологии [50].
Литература
- F. Sanger, S. Nicklen, A. R. Coulson. (1977). DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences. 74, 5463-5467;
- 12 методов в картинках: секвенирование нуклеиновых кислот;
- Криминалистика. Молекулярно-генетическая экспертиза;
- 454-секвенирование (высокопроизводительное пиросеквенирование ДНК);
- Код жизни: прочесть не значит понять;
- Новый взгляд на геном: не просто цепочка генов, а трехмерная сеть, интегрирующая функциональные домены ядра;
- Неуловимая архитектура хроматина мухи;
- Молекулы и эпигеном ;
- Загадочное путешествие некодирующей РНК Xist по X-хромосоме;
- Нанопоровое секвенирование: на пороге третьей геномной революции;
- Miten Jain, Sergey Koren, Karen H Miga, Josh Quick, Arthur C Rand, et. al.. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 36, 338-345;
- Mun Hua Tan, Christopher M Austin, Michael P Hammer, Yin Peng Lee, Laurence J Croft, Han Ming Gan. (2018). Finding Nemo: hybrid assembly with Oxford Nanopore and Illumina reads greatly improves the clownfish (Amphiprion ocellaris) genome assembly. GigaScience. 7;
- Kang Du, Matthias Stöck, Susanne Kneitz, Christophe Klopp, Joost M. Woltering, et. al.. (2020). The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat Ecol Evol. 4, 841-852;
- Геномная головоломка: открой в себе мозаика;
- Timour Baslan, James Hicks. (2017). Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 17, 557-569;
- Ethan J. Armand, Junhao Li, Fangming Xie, Chongyuan Luo, Eran A. Mukamel. (2021). Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron. 109, 11-26;
- Xianwen Ren, Guojie Zhong, Qiming Zhang, Lei Zhang, Yujie Sun, Zemin Zhang. (2020). Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 30, 763-778;
- Xiaoning Tang, Yongmei Huang, Jinli Lei, Hui Luo, Xiao Zhu. (2019). The single-cell sequencing: new developments and medical applications. Cell Biosci. 9;
- David Lähnemann, Johannes Köster, Ewa Szczurek, Davis J. McCarthy, Stephanie C. Hicks, et. al.. (2020). Eleven grand challenges in single-cell data science. Genome Biol. 21;
- Ping Hu, Wenhua Zhang, Hongbo Xin, Glenn Deng. (2016). Single Cell Isolation and Analysis. Front. Cell Dev. Biol.. 4;
- 12 методов в картинках: проточная цитофлуориметрия;
- Опухолевые разговоры, или Роль микроокружения в развитии рака;
- Peter See, Josephine Lum, Jinmiao Chen, Florent Ginhoux. (2018). A Single-Cell Sequencing Guide for Immunologists. Front. Immunol.. 9;
- Yong Hou, Kui Wu, Xulian Shi, Fuqiang Li, Luting Song, et. al.. (2015). Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. GigaSci. 4;
- R. Drmanac, A. B. Sparks, M. J. Callow, A. L. Halpern, N. L. Burns, et. al.. (2010). Human Genome Sequencing Using Unchained Base Reads on Self-Assembling DNA Nanoarrays. Science. 327, 78-81;
- Ana Conesa, Pedro Madrigal, Sonia Tarazona, David Gomez-Cabrero, Alejandra Cervera, et. al.. (2016). A survey of best practices for RNA-seq data analysis. Genome Biol. 17;
- Luwen Ning, Geng Liu, Guibo Li, Yong Hou, Yin Tong, Jiankui He. (2014). Current Challenges in the Bioinformatics of Single Cell Genomics. Front. Oncol.. 4;
- Fei Ji, Ruslan I. Sadreyev. (2019). Single‐Cell RNA‐seq: Introduction to Bioinformatics Analysis. Current Protocols in Molecular Biology. 127;
- Lisa N. Waylen, Hieu T. Nim, Luciano G. Martelotto, Mirana Ramialison. (2020). From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun Biol. 3;
- Michaela Asp, Joseph Bergenstråhle, Joakim Lundeberg. (2020). Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays. 42, 1900221;
- Eric Lubeck, Long Cai. (2012). Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods. 9, 743-748;
- #6 2. Новое решение Visium от 10x Genomics для анализа пространственной экспрессии генов. (2020). SkyGen;
- Samuel G. Rodriques, Robert R. Stickels, Aleksandrina Goeva, Carly A. Martin, Evan Murray, et. al.. (2019). Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 363, 1463-1467;
- Merritt C.R., Ong G.T., Church S., Barker K.B., Geiss G. et al. (2019). High multiplex, digital spatial profiling of proteins and RNA in fixed tissue using genomic detection methods. bioRxiv;
- Joseph Bergenstråhle, Ludvig Bergenstråhle, Joakim Lundeberg. (2020). SpatialCPie: an R/Bioconductor package for spatial transcriptomics cluster evaluation. BMC Bioinformatics. 21;
- Qianqian Song, Jing Su. (2021). DSTG: deconvoluting spatial transcriptomics data through graph-based artificial intelligence. Briefings in Bioinformatics;
- Ekaterina Khrameeva, Ilia Kurochkin, Dingding Han, Patricia Guijarro, Sabina Kanton, et. al.. (2020). Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res.. 30, 776-789;
- Ugomma C. Eze, Aparna Bhaduri, Maximilian Haeussler, Tomasz J. Nowakowski, Arnold R. Kriegstein. (2021). Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat Neurosci. 24, 584-594;
- Ethan J. Armand, Junhao Li, Fangming Xie, Chongyuan Luo, Eran A. Mukamel. (2021). Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron. 109, 11-26;
- W. Brad Ruzicka, Shahin Mohammadi, Jose Davila-Velderrain, Sivan Subburaju, Daniel Reed Tso, et. al. Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic axis and transcriptional resilience — Cold Spring Harbor Laboratory;
- Marta Olah, Vilas Menon, Naomi Habib, Mariko F. Taga, Yiyi Ma, et. al.. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun. 11;
- Hugo J.R. Fernandes, Nikolaos Patikas, Stefanie Foskolou, Sarah F. Field, Jong-Eun Park, et. al.. (2020). Single-Cell Transcriptomics of Parkinson’s Disease Human In Vitro Models Reveals Dopamine Neuron-Specific Stress Responses. Cell Reports. 33, 108263;
- Haide Chen, Fang Ye, Guoji Guo. (2019). Revolutionizing immunology with single-cell RNA sequencing. Cell Mol Immunol. 16, 242-249;
- Grégory Seumois, Ciro Ramírez-Suástegui, Benjamin J. Schmiedel, Shu Liang, Bjoern Peters, et. al.. (2020). Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci. Immunol.. 5, eaba6087;
- Анализ индивидуальных репертуаров Т-клеточных рецепторов;
- Т-лимфоциты: путешественники и домоседы;
- Тимоцитов ведут в зоопарк. Что такое эктопическая экспрессия и как она защищает нас от аутоиммунитета;
- Bora Lim, Yiyun Lin, Nicholas Navin. (2020). Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell. 37, 456-470;
- Jian Liu, Tianmin Xu, Yuemei Jin, Bingyu Huang, Yan Zhang. (2021). Progress and Clinical Application of Single-Cell Transcriptional Sequencing Technology in Cancer Research. Front. Oncol.. 10;
- Tanja Woyke, Devin F R Doud, Frederik Schulz. (2017). The trajectory of microbial single-cell sequencing. Nat Methods. 14, 1045-1054;
- Fang Ye, Wentao Huang, Guoji Guo. (2017). Studying hematopoiesis using single-cell technologies. J Hematol Oncol. 10;
- Mansi Verma, Samarth Kulshrestha, Ayush Puri. (2017). Genome Sequencing. Methods in Molecular Biology. 3-33;
- Kristen R. Maynard, Leonardo Collado-Torres, Lukas M. Weber, Cedric Uytingco, Brianna K. Barry, et. al.. (2021). Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci. 24, 425-436.



Комментарии
0Чтобы оставить комментарий, необходимо
войти